Classification rationale
PM2
BP4BP6
Likely Benign
ETV6 c.329-15C>A
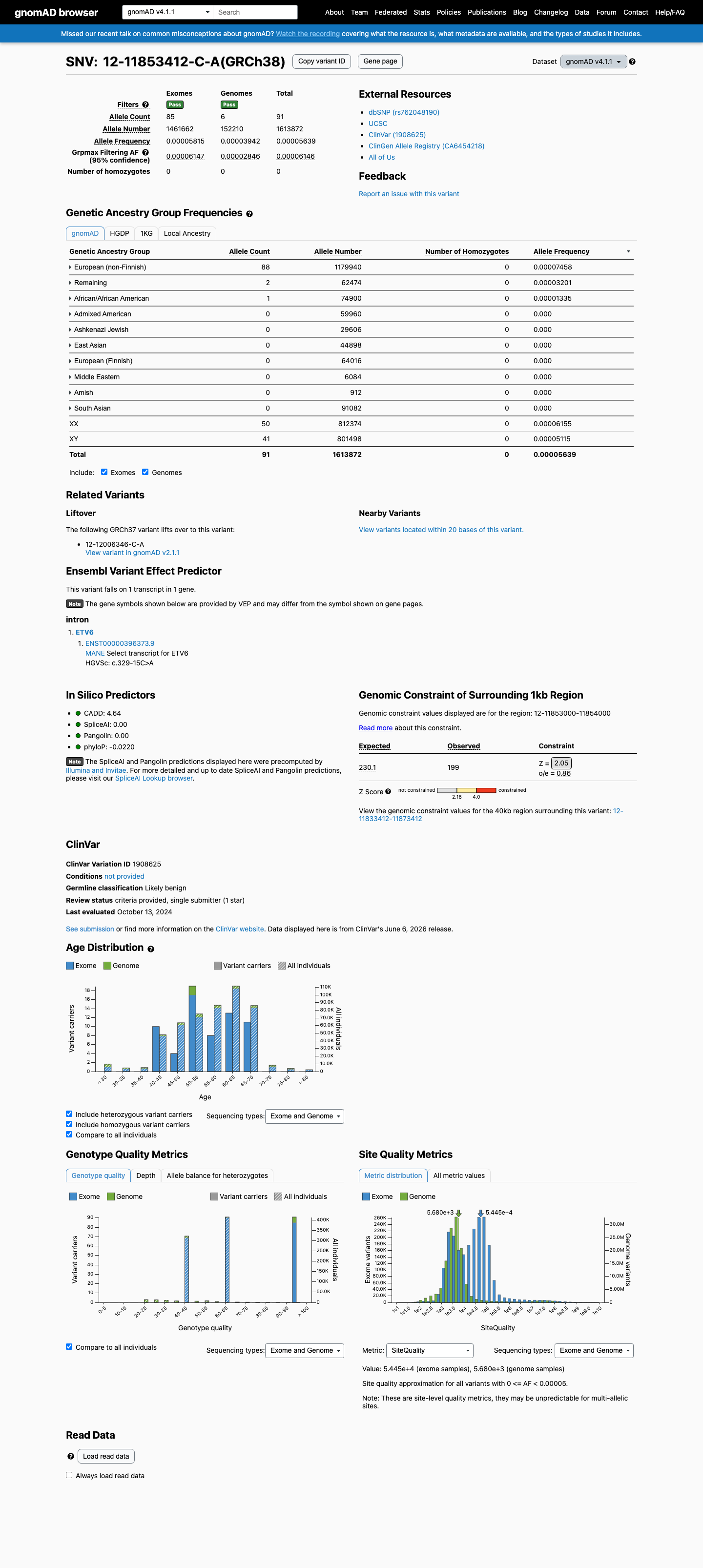
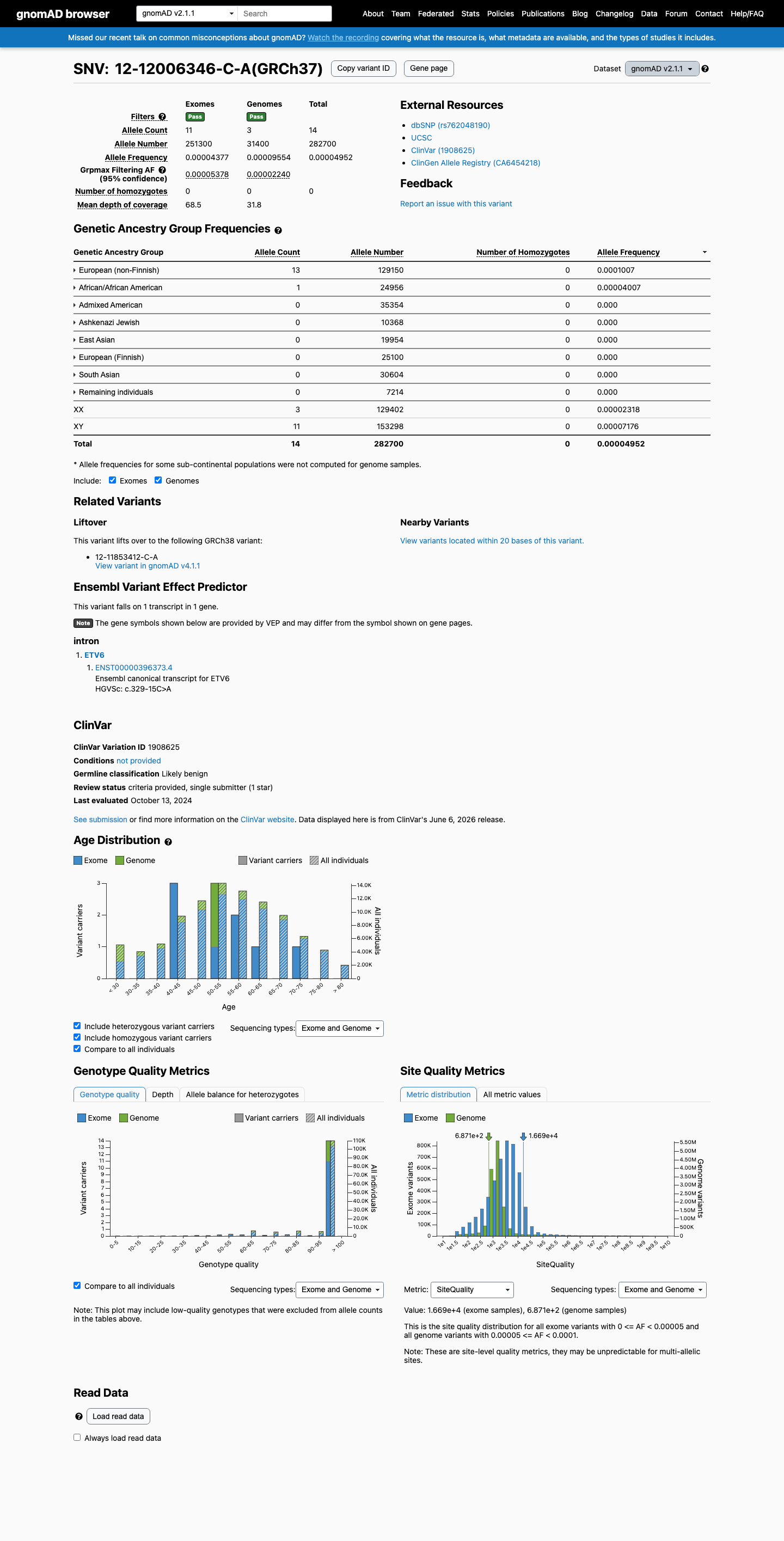
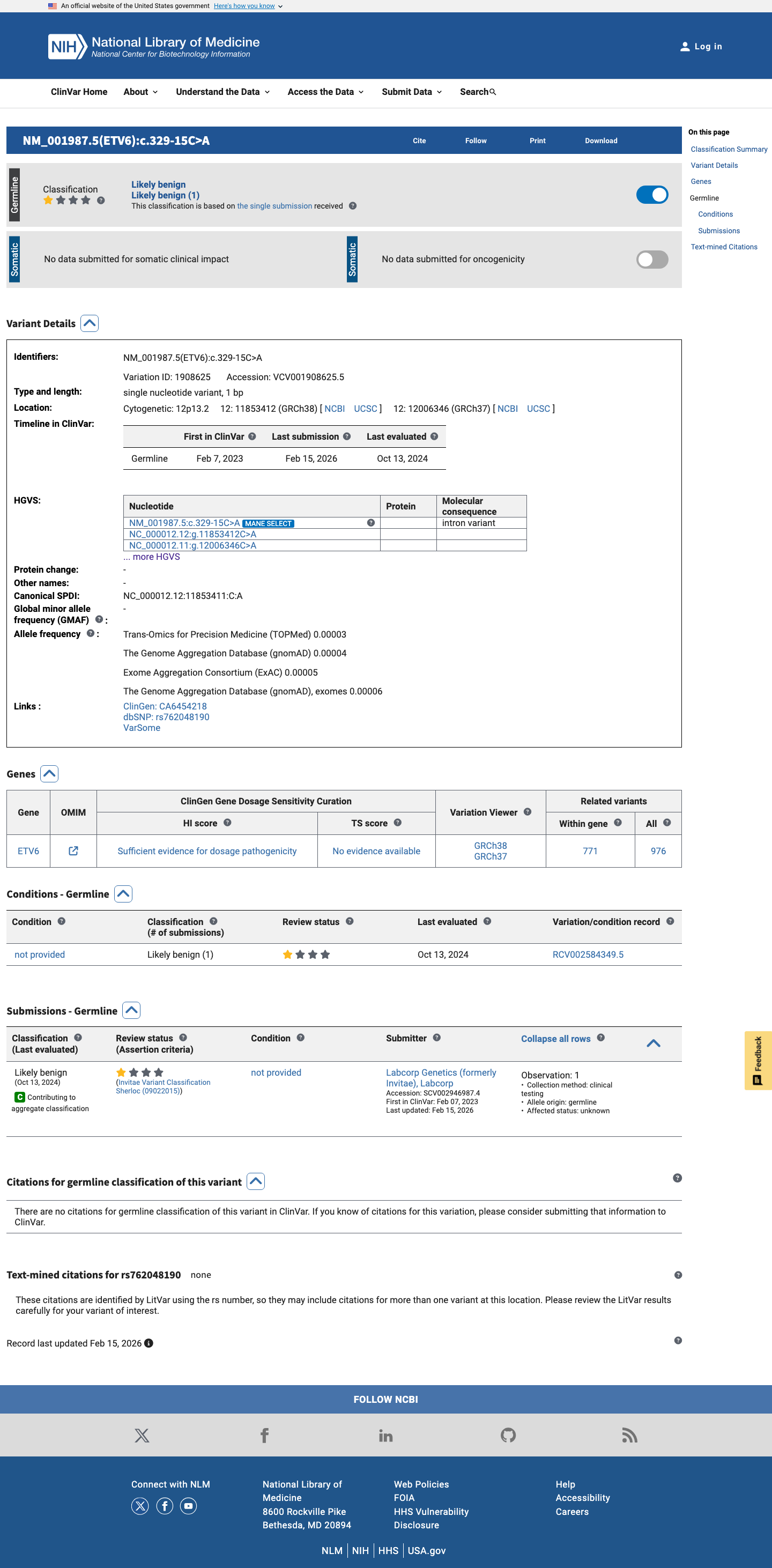
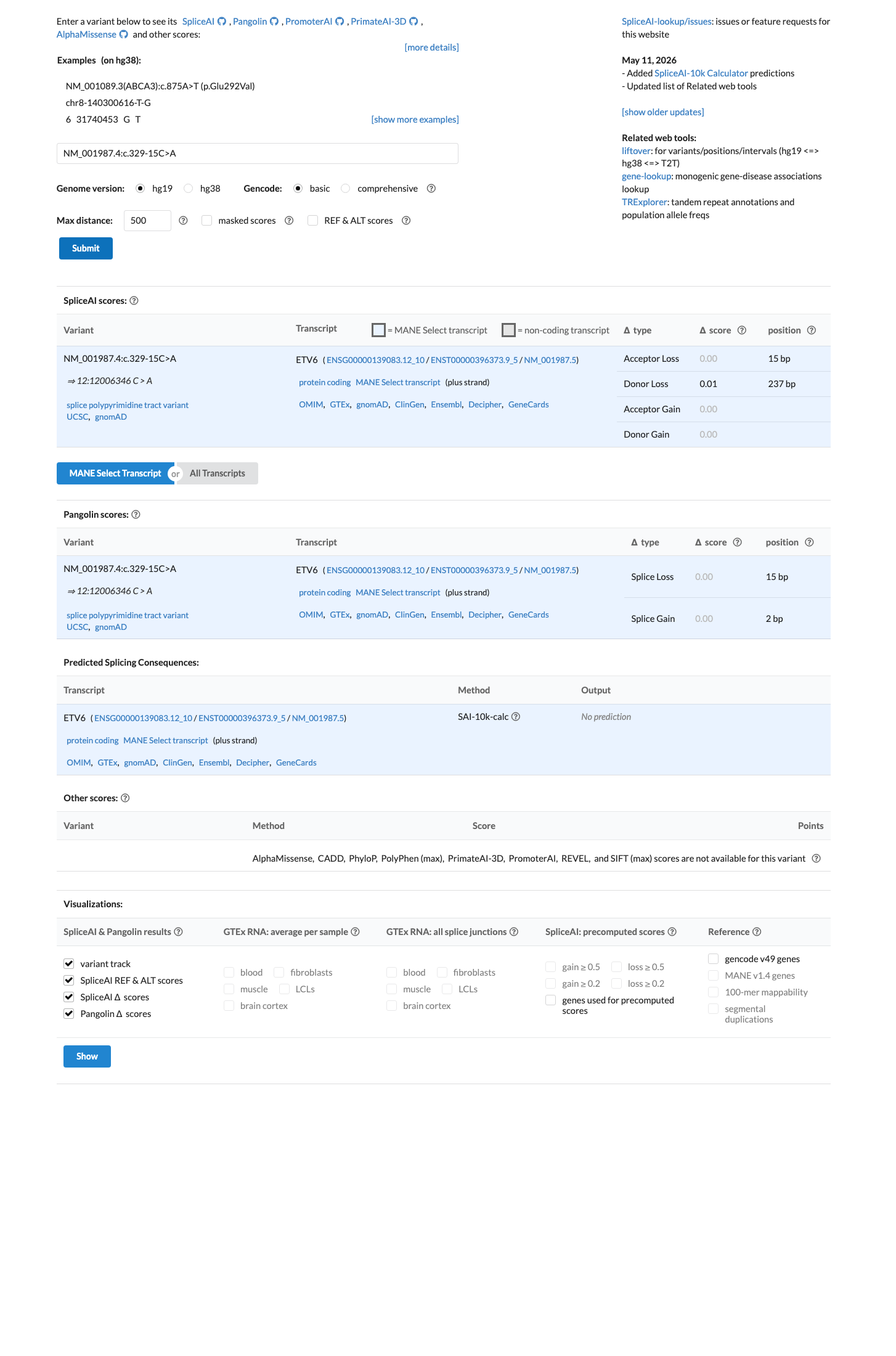
NM_001987.4:c.329-15C>A is an intronic substitution in ETV6 (intron 3, c.329-15) with no predicted splicing impact (SpliceAI max delta 0.01).1 This variant is present in gnomAD at very low frequency (v2.1: 14/282,700 alleles, AF=0.00495%; v4.1: 91/1,613,872 alleles, AF=0.00564%) with no homozygotes observed.2 ClinVar reports this variant as Likely benign by Labcorp Genetics/Invitae (ClinVarID 1908625, SCV002946987), with criteria provided.3 In silico evidence supports a benign interpretation: SpliceAI predicts no splicing alteration, and no computational tool suggests a deleterious effect.4
PM2 + BP4 + BP6
→
Likely Benign