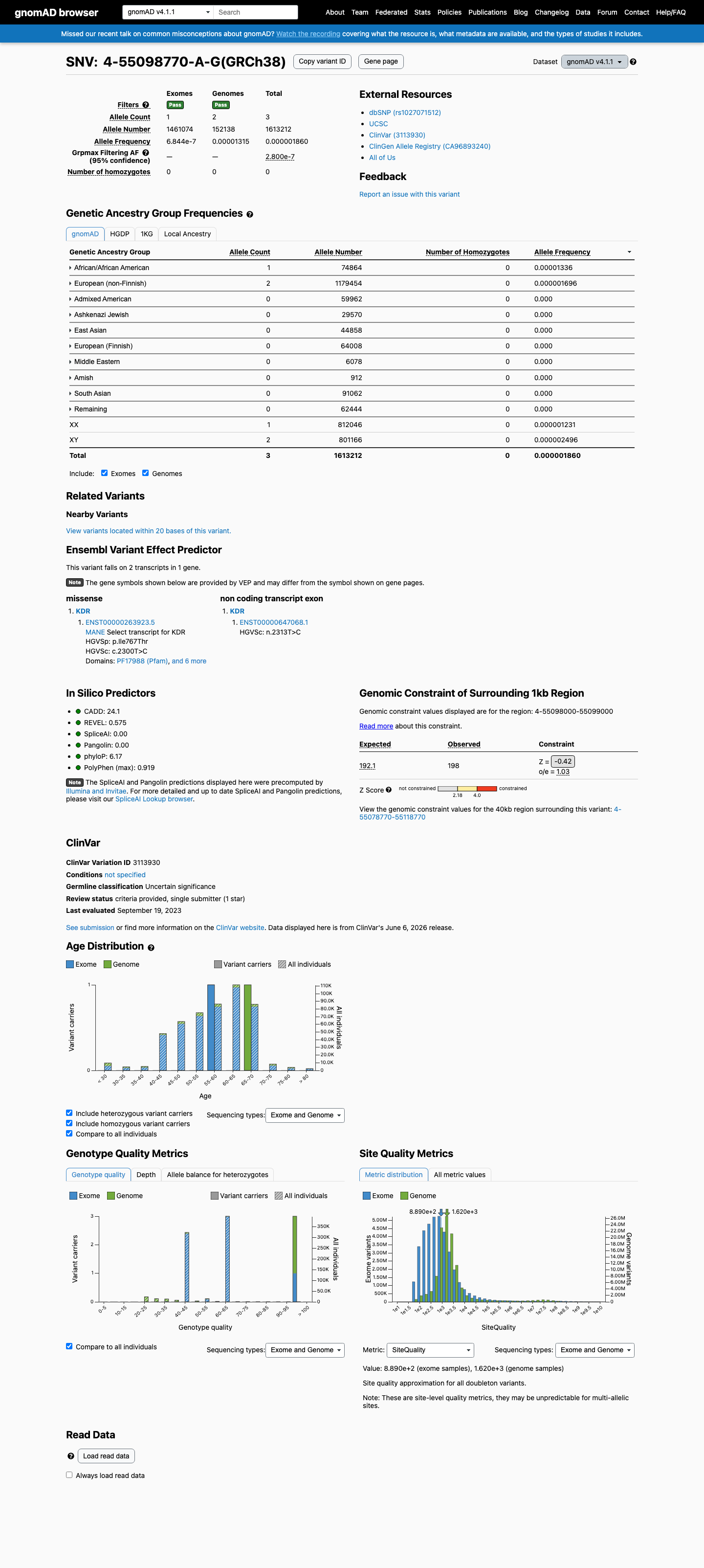
NM_002253.3:c.2300T>C (p.Ile767Thr) is a missense variant in the KDR gene, located in the transmembrane domain. This variant is absent from gnomAD v2.1 and is extremely rare in gnomAD v4.1 (allele frequency 1.86e-06, 3/1,613,212 alleles), meeting PM2 at moderate strength.1 The variant is absent from gnomAD-Canada v1.0 and not reported in COSMIC. In silico predictions are equivocal: SpliceAI predicts no splice impact (max delta 0.00), REVEL score is 0.575 (borderline), and BayesDel is -0.004 (neutral). Neither PP3 nor BP4 is met.2 The variant has been reported in ClinVar as Uncertain significance by a single clinical laboratory (Ambry Genetics). No submitter has classified it as pathogenic or benign.3 OncoKB classifies KDR I767T as Unknown Oncogenic Effect with no curated functional evidence. No published functional studies or case-level data specific to this variant were identified.4 No CSPEC or VCEP framework exists for KDR. Assessment follows generic ACMG/AMP 2015 guidelines.5 The single met criterion is PM2 (moderate). No pathogenic criteria at supporting strength or higher are met beyond PM2. Insufficient evidence is available to reach a likely pathogenic or pathogenic classification. Insufficient benign evidence is available to reach a likely benign or benign classification. The variant remains a variant of uncertain significance.
KDR
Final classification
VUS
KDR c.2300T>C · p.Ile767Thr
KDR
NM_002253.3:c.2300T>C (p.Ile767Thr) is a missense variant in the KDR gene, located in the transmembrane domain.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 moderate; combination = 1 moderate, which maps to VUS.
Classification rationale
PM2
VUS
KDR c.2300T>C
PM2
→
VUS
Gene diagram
· NM_002253.3 · variants mapped to exon structure
KDR
NM_002253.3
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 22 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
moderate
Pathogenic
This variant is absent from gnomAD v2.1 and is extremely rare in gnomAD v4.1 (allele frequency 1.86e-06, 3/1,613,212 alleles, 0.00019%), well below the 0.1% threshold for PM2. It is also absent from gnomAD-Canada v1.0. The grpmax filtering allele frequency is 2.8e-07.
Absent from gnomAD v2.1.gnomAD v4.1: AF 1.86e-06 (3/1613
Assessed · not applied
Pathogenic
PS1
No evidence of a different pathogenic nucleotide change at the same amino acid position (p.Ile767) was identified in ClinVar or the literature.
PS2
No de novo occurrence data with confirmed paternity and maternity are available for this variant.
PS3
No well-established in vitro or in vivo functional studies were identified for this variant.
PS4
No case-control studies or prevalence comparisons in affected vs.
PM1
The variant p.Ile767Thr is located in the transmembrane domain of KDR (VEGFR2), not in a statistically significant mutational hotspot.
PM5
No pathogenic missense variant at the same amino acid residue (p.Ile767) was identified in ClinVar or published literature to serve as a PM5 comparator.
PM6
No de novo occurrence data (with or without confirmed parentage) are available for this variant.
PP1
No cosegregation data in affected family members are available for this variant.
PP2
Insufficient data to determine whether KDR has a low rate of benign missense variation and whether missense variants are a common mechanism of disease.
PP3
Multiple lines of computational evidence do not support a deleterious effect.
PP4
No proband phenotype or family history information is available to assess whether the clinical presentation is highly specific for KDR-related disease.
PP5
No reputable source has reported this variant as pathogenic.
Benign
BA1
The allele frequency in gnomAD v4.1 is 1.86e-06 (0.00019%), far below the 1% BA1 threshold for non-VCEP frameworks.
BS1
The allele frequency in gnomAD v4.1 is 1.86e-06 (0.00019%), far below the 0.3% BS1 threshold for non-VCEP frameworks.
BS2
No data on observation of this variant in healthy adult individuals are available to assess BS2.
BS3
No well-established in vitro or in vivo functional studies showing no damaging effect on protein function or splicing are available for this variant.
BS4
No family-based cosegregation data are available to assess lack of segregation in affected family members.
BP1
While KDR has literature-level support for loss-of-function as a disease mechanism in germline predisposition (familial Hodgkin lymphoma), there is insufficient evidence that KDR-related disease is caused primarily by truncating variants as opposed to missense variants.
BP2
No data on observation of this variant in trans with a pathogenic variant in KDR or in cis with a pathogenic variant are available.
BP4
Multiple lines of computational evidence do not consistently suggest no impact.
BP5
No data are available identifying an alternate molecular basis for disease in a proband carrying this variant.
BP6
No reputable source has reported this variant as benign.
N/A · 5
PVS1 · PM3 · PM4 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 1.85964e-06; MAF= 0.00019%, 3/1613212 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 1.33576e-05; MAF= 0.00134%, 1/74864 alleles, homozygotes = 0); grpmax FAF= 2.8e-07.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00019%
· 3 / 1,613,212
0 hom · FAF 2.8e-05%
0 hom · FAF 2.8e-05%
African/African American 1 / 74,864 |
0.0013% |
European (non-Finnish) 2 / 1,179,454 |
0.00017% |
+ 8 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
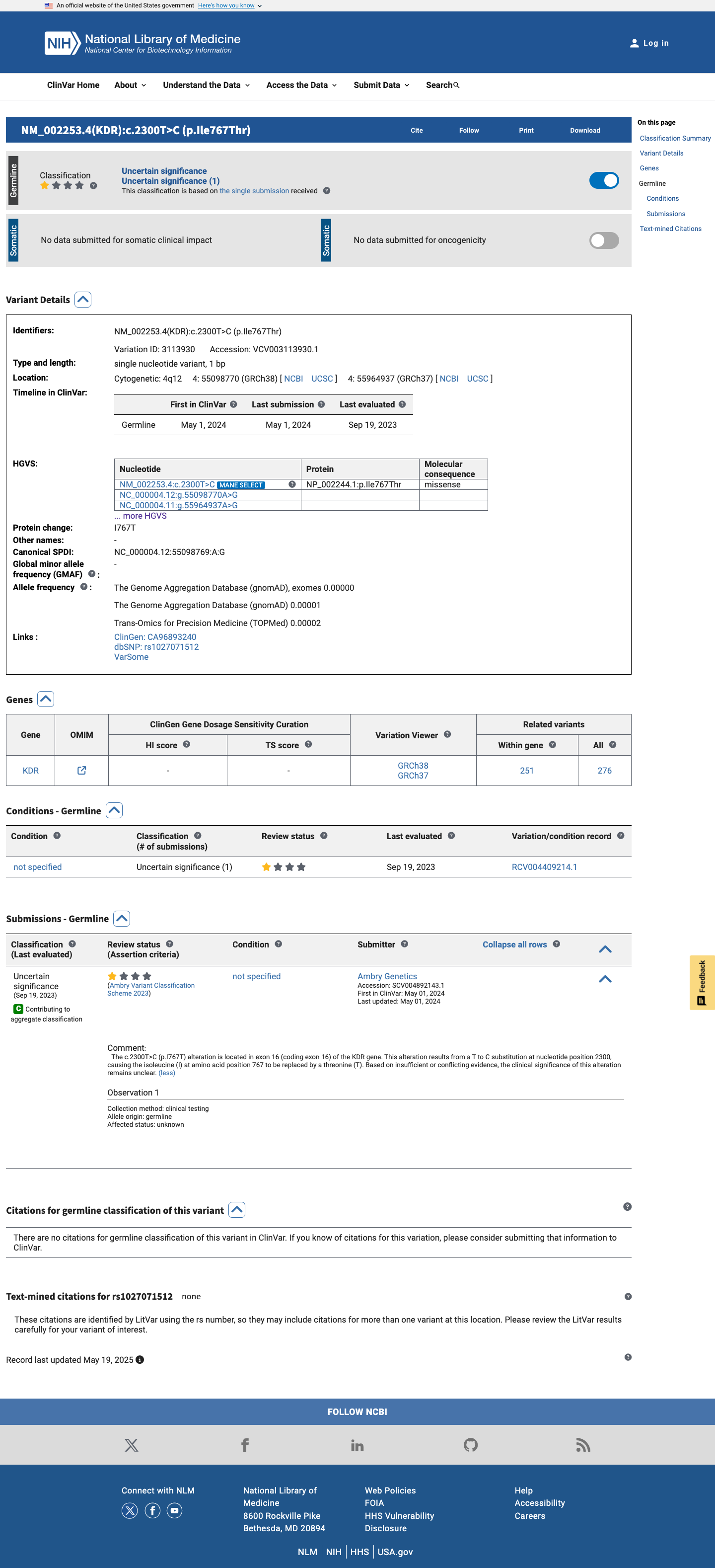
This variant has been reported in ClinVar as Uncertain significance (1 clinical laboratory). (ClinVarID = 3113930)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.575. BayesDel score = -0.00416777.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. KDR, a receptor tyrosine kinase, is altered by mutation or amplification in various cancers, most frequently in skin cancers.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links