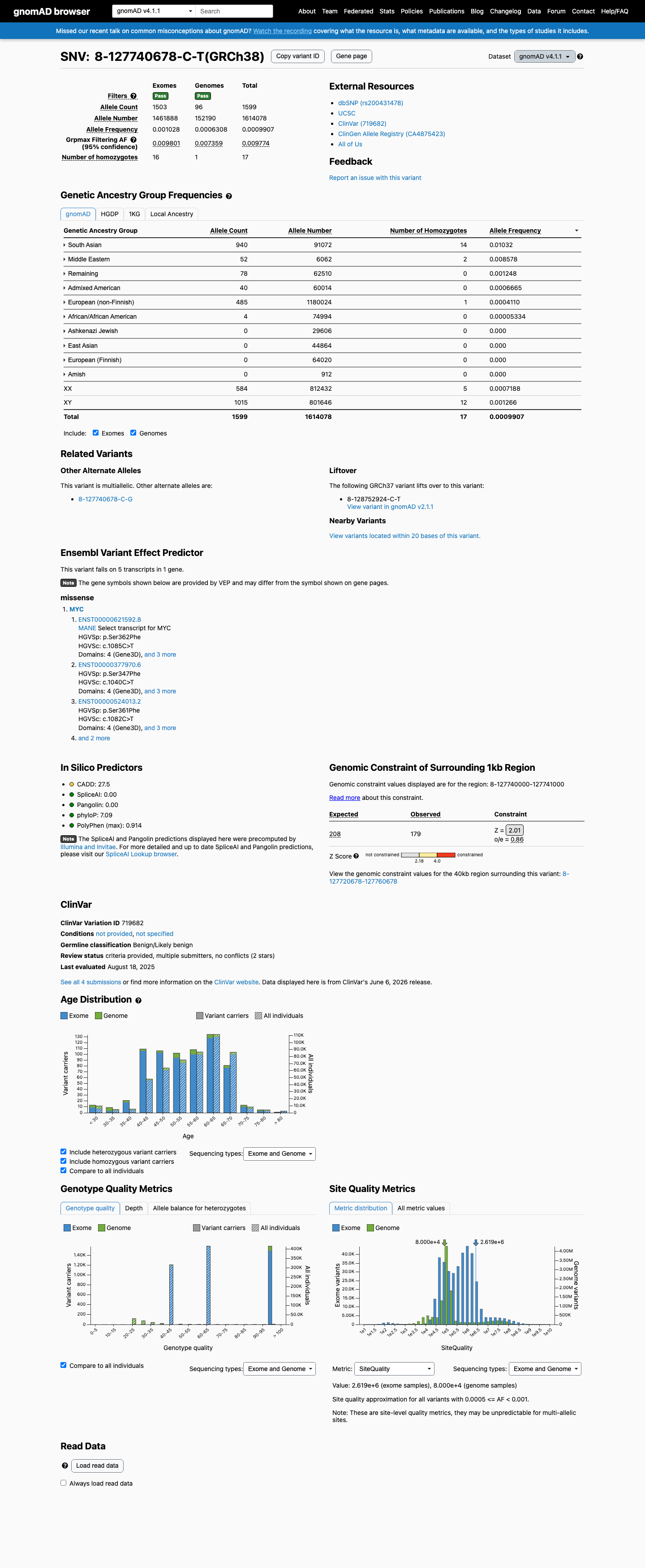
NM_002467.6:c.1085C>T (p.Ser362Phe) is present at a grpmax filtering allele frequency of 0.977% in the South Asian population of gnomAD v4.1, exceeding the BS1 threshold of >0.3%, which indicates the variant is too common to be a fully penetrant pathogenic allele for a rare Mendelian disorder.1 This variant has been observed in 17 homozygous individuals in gnomAD v4.1, which is incompatible with a fully penetrant early-onset dominant disorder, meeting BS2 at strong benign strength.2 Multiple computational predictors indicate a benign effect: REVEL score 0.445 (below 0.5 threshold), BayesDel −0.151 (benign-leaning), and SpliceAI max delta 0.00 (no predicted splicing impact), supporting BP4.3 ClinVar reports this variant as Likely benign by two clinical laboratories and Benign by one clinical laboratory (Variation ID 719682), with criteria provided, supporting BP6.4
MYC
Final classification
Benign
MYC c.1085C>T · p.Ser362Phe
MYC
NM_002467.6:c.1085C>T (p.Ser362Phe) is present at a grpmax filtering allele frequency of 0.977% in the South Asian population of gnomAD v4.1, exceeding the BS1 threshold of >0.3%, which indicates the variant is too common to be a fully penetrant pathogenic allele for a rare Mendelian disorder.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS1 strong, BS2 strong, BP4 supporting, BP6 supporting; combination = 2 strong benign + 2 supporting benign, which maps to Benign.
Classification rationale
BS1BS2BP4BP6
Benign
MYC c.1085C>T
BS1 + BS2 + BP4 + BP6
→
Benign
Gene diagram
· NM_002467.6 · variants mapped to exon structure
MYC
NM_002467.6
Fetching transcript structure from UCSC…
Applied criteria · 4 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000990658; MAF= 0.09907%, 1599/1614078 alleles, homozygotes = 17) and has highest observed frequency in the South Asian population (AF= 0.0103215; MAF= 1.03215%, 940/91072 alleles, homozygotes = 14); grpmax FAF= 0.00977366.
v2.1
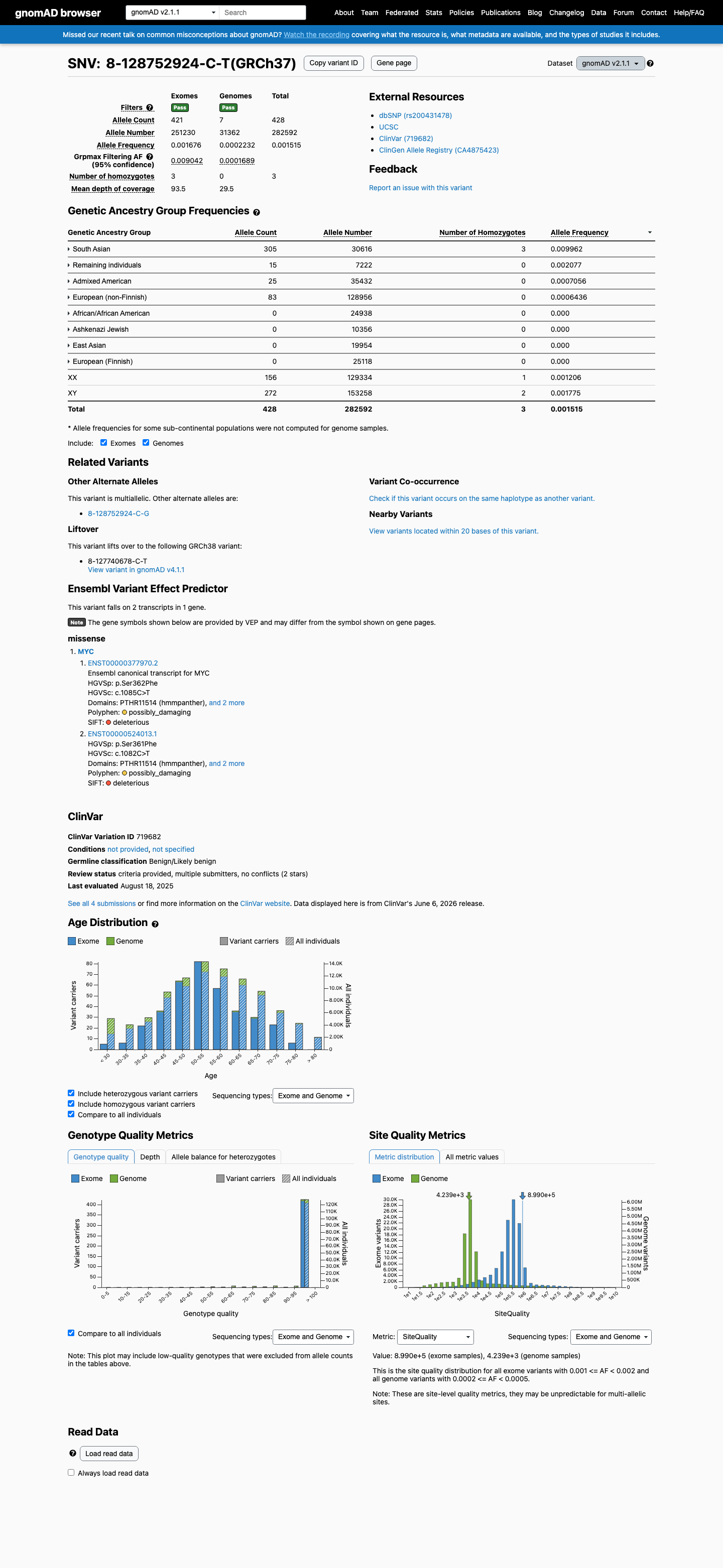
This variant is present in gnomAD v2.1 (AF= 0.00151455; MAF= 0.15146%, 428/282592 alleles, homozygotes = 3) and has highest observed frequency in the South Asian population (AF= 0.00996211; MAF= 0.99621%, 305/30616 alleles, homozygotes = 3); grpmax FAF= 0.00904243.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0013572204125950054, 25/18420 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.099%
· 1599 / 1,614,078
17 hom · FAF 0.98%
17 hom · FAF 0.98%
South Asian 940 / 91,072 |
1% 14 hom |
Middle Eastern 52 / 6,062 |
0.86% 2 hom |
Remaining individuals 78 / 62,510 |
0.12% |
Admixed American 40 / 60,014 |
0.067% |
European (non-Finnish) 485 / 1,180,024 |
0.041% 1 hom |
African/African American 4 / 74,994 |
0.0053% |
+ 4 not observed (European (Finnish), Amish, East Asian, Ashkenazi Jewish)
gnomAD v2.1
0.15%
· 428 / 282,592
3 hom · FAF 0.9%
3 hom · FAF 0.9%
South Asian 305 / 30,616 |
1% 3 hom |
Remaining individuals 15 / 7,222 |
0.21% |
Admixed American 25 / 35,432 |
0.071% |
European (non-Finnish) 83 / 128,956 |
0.064% |
+ 4 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish))
gnomAD Canada 🇨🇦
0.14%
· 25 / 18,420
0 hom · FAF 0.45%
0 hom · FAF 0.45%
South Asian 11 / 1,362 |
0.81% |
Remaining individuals 4 / 1,138 |
0.35% |
East Asian 3 / 1,338 |
0.22% |
Latino/Admixed American 1 / 838 |
0.12% |
European (non-Finnish) 6 / 11,740 |
0.051% |
+ 4 not observed (African/African American, Ashkenazi Jewish, European (Finnish), Middle Eastern)
ClinVar
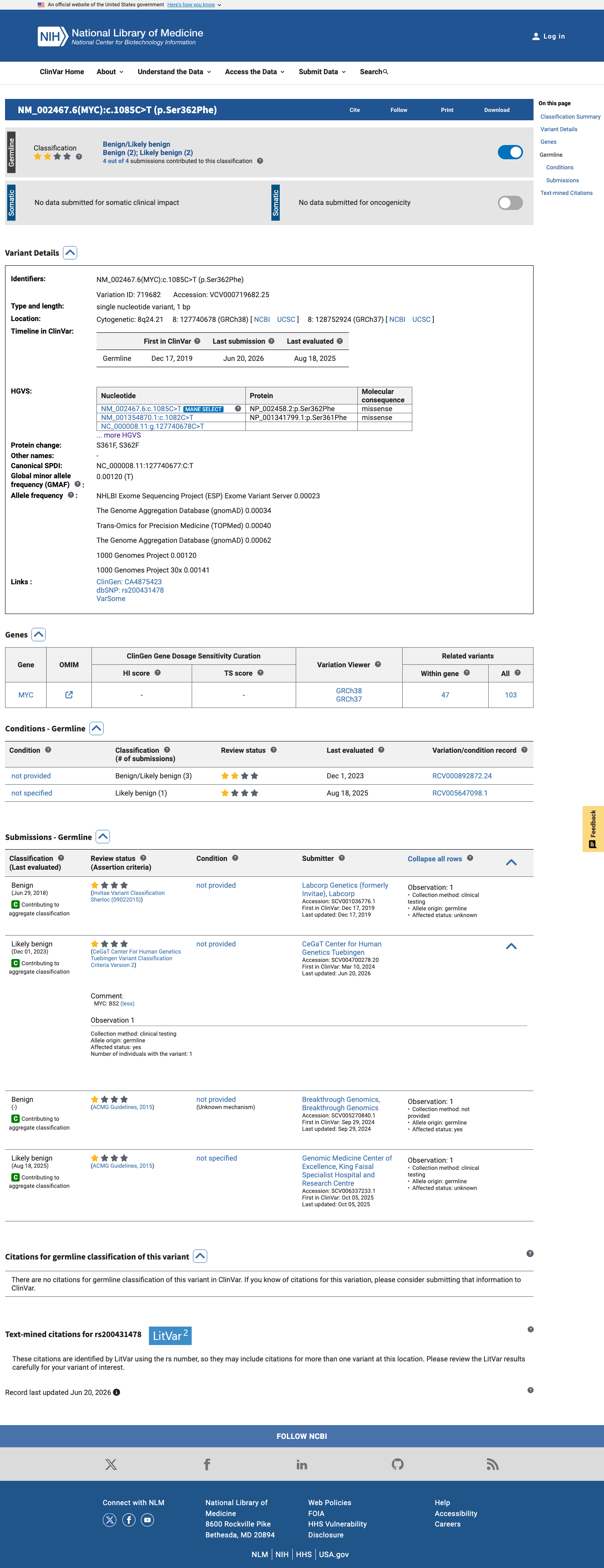
This variant has been reported in ClinVar as Likely benign (2 clinical laboratories) and as Benign (1 clinical laboratory). (ClinVarID = 719682)
In silico
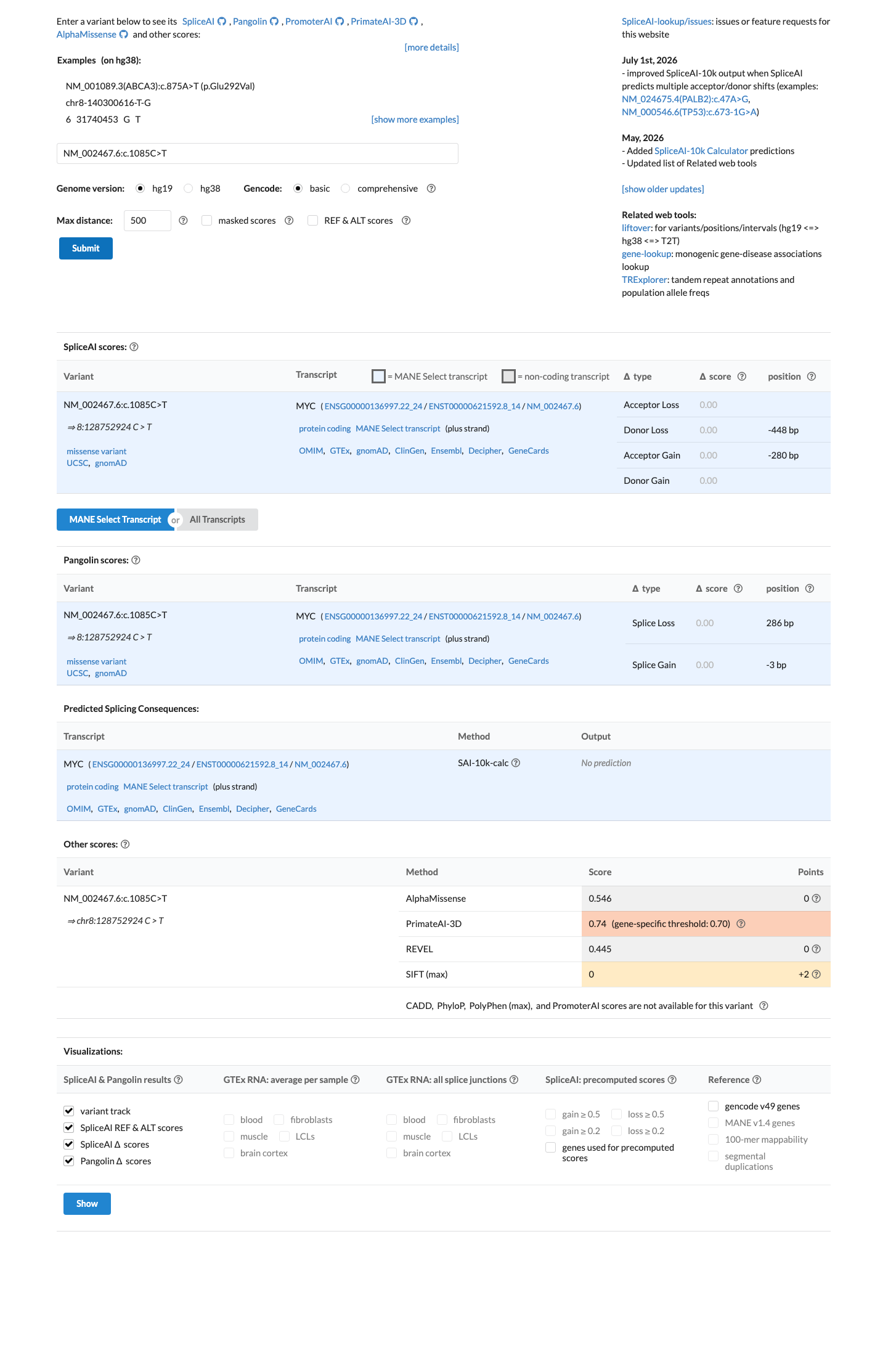
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.445. BayesDel score = -0.151424.
Functional
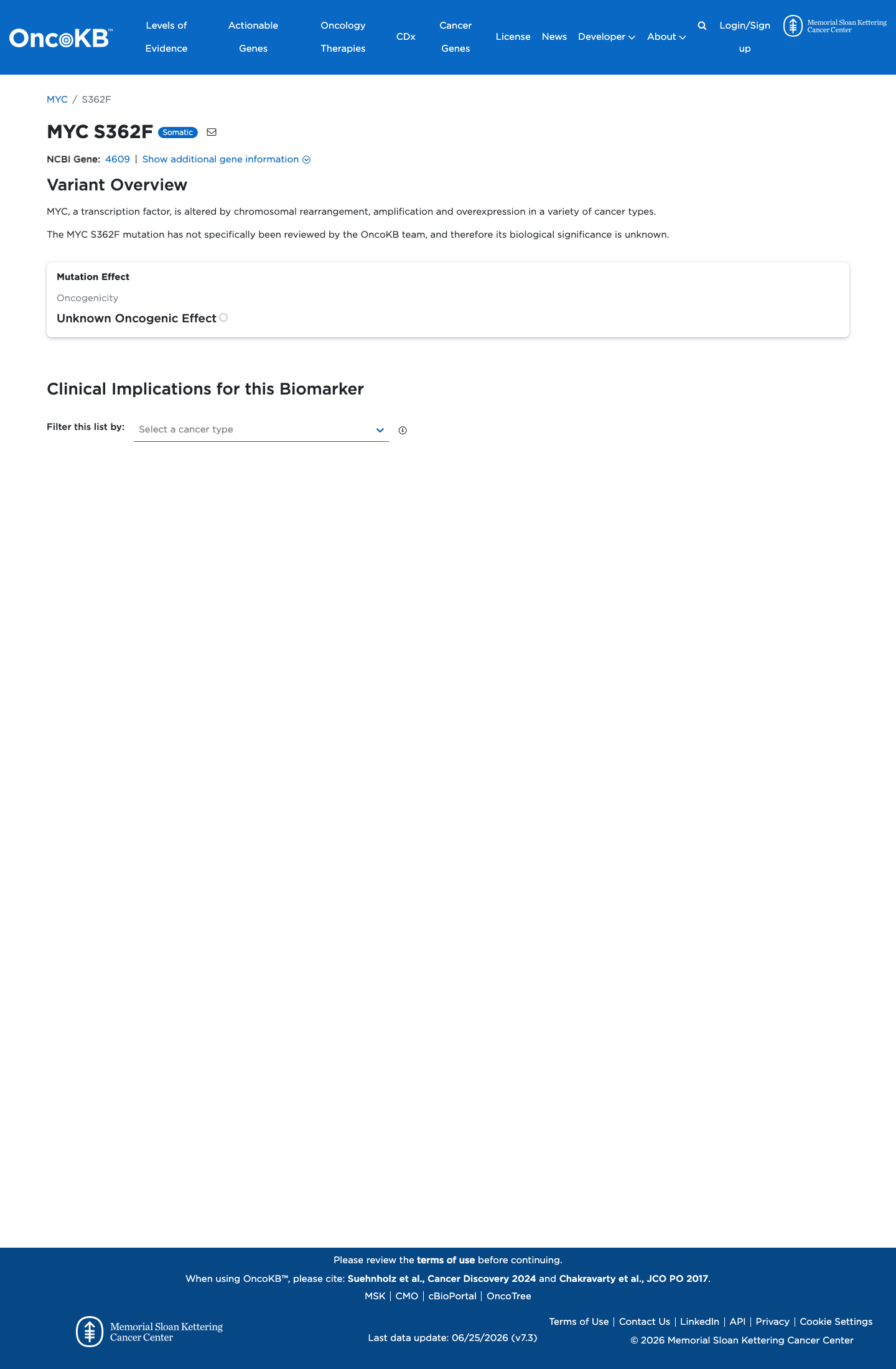
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. MYC, a transcription factor, is altered by chromosomal rearrangement, amplification and overexpression in a variety of cancer types.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
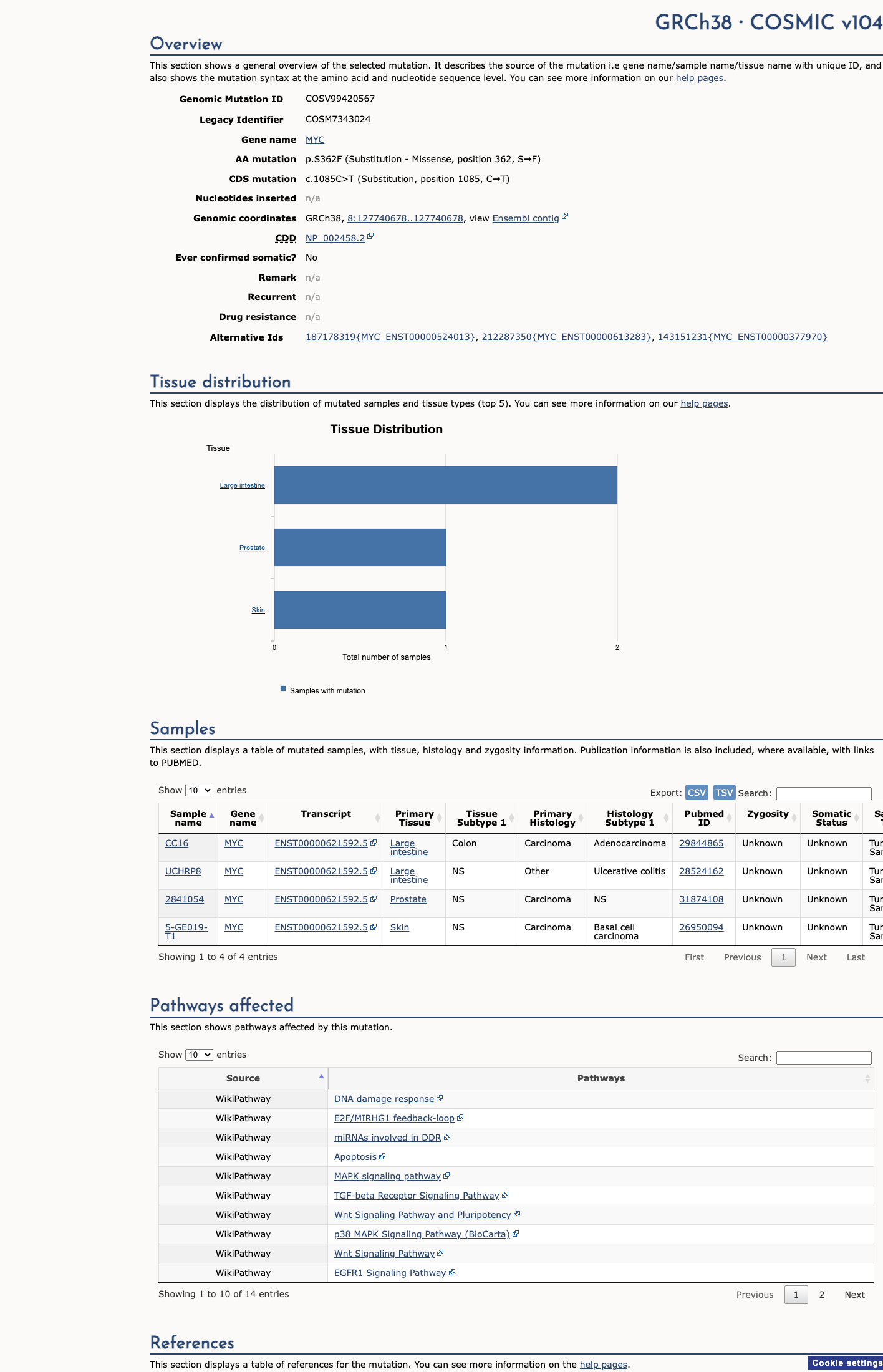
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV99420567, n = 4 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 2 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR