PVS1 (very strong): NM_002485.4:c.657_661del is a frameshift deletion introducing a premature termination codon (p.Lys219AsnfsTer16) in exon 6 of 16, predicting NMD. NBN loss of function is a well-established mechanism for Nijmegen breakage syndrome.1 PS3 (moderate): Functional characterization demonstrated the 657del5 allele produces a truncated p26 fragment lacking the MRE11 interaction domain and a partially functional p70 fragment via internal translation initiation; NBS cells exhibit radioresistant DNA synthesis, indicating impaired S-phase checkpoint.2 PS4 (strong): The variant is highly enriched in affected individuals. Approximately 90% of NBS patients are homozygous for this allele. Case-control studies demonstrate significantly elevated odds ratios: familial prostate cancer OR=16 (P<0.0001), medulloblastoma OR=4.86 (P=0.0028).3 PM1 (supporting): The frameshift truncation at codon 219 removes the MRE11 interaction domain (aa 433-754), a critical functional domain for DNA double-strand break repair complex formation.4 PM2 (supporting): Extremely low population frequency (gnomAD v2.1 AF=0.0202%, v4.1 AF=0.0225%), no homozygotes observed, and absent from gnomAD-Canada.5 PP1 (moderate): Co-segregation demonstrated in multiple families for both NBS (recessive) and cancer predisposition, with LOH of wild-type allele in 7/8 prostate tumors from carriers.6 PP5 (supporting): Classified as Pathogenic by 38 clinical laboratories in ClinVar (Variation ID 6940).7 The overall classification supports Pathogenic using generic ACMG/AMP 2015 combination rules: 1 very strong (PVS1) + 1 strong (PS4) + 1 moderate (PS3) + 1 moderate (PP1) + 3 supporting (PM1, PM2, PP5) far exceeds the threshold for Pathogenic classification.8
NBN
Final classification
Pathogenic
NBN c.657_661del · p.Lys219AsnfsTer16
NBN
PVS1 (very strong): NM_002485.4:c.657_661del is a frameshift deletion introducing a premature termination codon (p.Lys219AsnfsTer16) in exon 6 of 16, predicting NMD. NBN loss of function is a well-established mechanism for Nijmegen breakage syndrome.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 very strong, PS3 moderate, PS4 strong, PM1 supporting, PM2 supporting, PP1 moderate, PP5 supporting; combination = 1 very strong + 1 strong + 2 moderate + 3 supporting, which maps to Pathogenic.
Classification rationale
PVS1PS3PS4PM1PM2PP1PP5
Pathogenic
NBN c.657_661del
PVS1 + PS3 + PS4 + PM1 + PM2 + PP1 + PP5
→
Pathogenic
Gene diagram
· NM_002485.4 · variants mapped to exon structure
NBN
NM_002485.4
Fetching transcript structure from UCSC…
Applied criteria · 7 applied · 11 assessed
Applied · 7
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
NM_002485.4:c.657_661del is a frameshift deletion in exon 6 of 16 that introduces a premature termination codon at position 234 (p.Lys219AsnfsTer16), well upstream of the final exon-exon junction, predicting nonsense-mediated decay. NBN loss of function is a well-established mechanism for Nijmegen breakage syndrome (autosomal recessive). Under PMC6185798 generic PVS1 framework, frameshift variants in LoF-eligible genes warrant PVS1 at full strength. Although internal translation initiation can produce a partially functional p70 fragment retaining the MRE11 interaction domain (PMID:11279524), this does not rescue NMD of the primary transcript and the allele remains disease-causing when homozygous.
Frameshift deletion c.657_661del in exon 6/16 creates PTC at codon 234predicting NMDNBN is a well-established germline LoF disease gene (Nijmegen breakage syndrome
✓
PS3
moderate
Pathogenic
Functional characterization of the 657del5 NBS1 allele by Maser et al. (PMID:11279524) demonstrated that the variant produces two distinct protein products: a 26-kD N-terminal fragment (NBS1p26) containing FHA/BRCT domains but lacking the MRE11 interaction domain, and a 70-kD protein (NBS1p70) generated via internal translation initiation that retains the MRE11 interaction domain but lacks the N-terminal FHA/BRCT domains. NBS1p70 partially restores MRE11 complex nuclear localization but does not confer full function; NBS 657del5 LCLs exhibit radioresistant DNA synthesis. This is a single study with variant-specific functional data qualifying for moderate-strength PS3.
Direct protein characterization of 657del5 allele: NBS1p26 lacks MRE11 interactionNBS1p70 retains MRE11 interaction but lacks FHA/BRCT domainsNBS1p70 partially restores MRE11 complex nuclear localization but fails to restore S-phase checkpoint (radioresistant DNA synthesis)
✓
PS4
strong
Pathogenic
The c.657_661del variant is highly enriched in affected individuals across multiple independent studies. In recessive NBS, approximately 90% of patients are homozygous for this founder allele (PMID:10799436, PMID:11093281). In heterozygous cancer risk, Cybulski et al. (PMID:14973119) reported OR=16 (P<0.0001) for familial prostate cancer and OR=3.9 (P=0.01) for nonfamilial prostate cancer versus 0.6% population controls. Ciara et al. (PMID:19908051) reported OR=4.86 (P=0.0028) for medulloblastoma. The variant is absent from gnomAD-Canada and at very low frequency globally (AF ~0.02%), further supporting strong enrichment in disease cohorts.
90% of NBS patients homozygous for 657del5 founder allele (PMID:10799436PMID:11093281)Familial prostate cancer OR=16
✓
PM1
supporting
Pathogenic
The c.657_661del frameshift truncates nibrin at codon 219, removing the MRE11 interaction domain (aa 433-754) from the primary p26 protein product. The MRE11 interaction domain is a well-characterized critical functional domain essential for DNA double-strand break repair complex formation. Although internal translation initiation produces a p70 fragment retaining this domain, the frameshift removes the domain from the canonical protein product.
Truncation at codon 219 removes MRE11 interaction domain (aa 433-754) from primary protein productMRE11 interaction domain is critical for DSB repair complex formationDomain disruption is supported by functional characterization in PMID:11279524
✓
PM2
supporting
Pathogenic
NM_002485.4:c.657_661del is present at very low frequency in population databases: gnomAD v2.1 AF=0.000202 (0.0202%, 57/282132 alleles), gnomAD v4.1 AF=0.000225 (0.0225%, 363/1613090 alleles), grpmax FAF=0.000297. No homozygotes observed. Absent from gnomAD-Canada. The frequency falls below the 0.1% PM2 threshold for a rare variant.
gnomAD v2.1 AF=0.0202% (57/282132)no homozygotesgnomAD v4.1 AF=0.0225% (363/1613090)
✓
PP1
moderate
Pathogenic
Co-segregation of c.657_661del with disease has been demonstrated in multiple families. In the NBS registry study (PMID:10799436), multiple sibships showed homozygous affected individuals (e.g., patients 1-2, 3-4-5-6, 9-10). In prostate cancer families (PMID:14973119), the 657del5 mutation was present in both affected males in each of 4 tested families, and LOH of the wild-type allele was observed in 7/8 prostate tumors from carriers. In medulloblastoma (PMID:19908051), heterozygous carriers were identified in 7/104 patients.
Multiple NBS sibships with co-segregation of homozygous 657del5 and disease (PMID:10799436)4 prostate cancer families with co-segregation in affected males (PMID:14973119)LOH of wild-type allele in 7/8 tumors from carriers
✓
PP5
supporting
Pathogenic
NM_002485.4:c.657_661del is classified as Pathogenic in ClinVar (Variation ID 6940) by 38 clinical laboratories. While the review status is criteria provided, single submitter, the 38-lab consensus with consistent Pathogenic classification across multiple independent clinical laboratories constitutes reputable source evidence.
ClinVar Variation ID 6940: Pathogenic by 38 clinical laboratoriesConsistent classification across independent clinical laboratories1 submitter classifies as risk factor
Assessed · not applied
Pathogenic
PS2
No de novo occurrence data was identified for NM_002485.4:c.657_661del in any reviewed publication.
PM6
No de novo occurrence of NM_002485.4:c.657_661del was identified in any reviewed publication.
PP3
Multiple lines of computational evidence do not support a damaging effect.
PP4
PP4 requires the specific patient's phenotype or family history to be highly specific for a disease with a single genetic etiology.
Benign
BA1
Allele frequency is far below the BA1 threshold.
BS1
Allele frequency is below the BS1 threshold.
BS3
Well-established functional studies do not show a benign effect.
BS4
Lack of segregation is not observed.
BP2
BP2 requires observation of the variant in trans with a pathogenic variant for a dominant disorder, or in cis with a pathogenic variant.
BP5
BP5 requires identification of the variant in a case with an alternate molecular basis for disease.
BP6
BP6 applies when a reputable source reports the variant as benign.
N/A · 9
PS1 · PM4 · PM5 · PP2 · BS2 · BP1 · BP3 · BP4 · BP7
Research & evidence
Population frequency · supports pathogenic
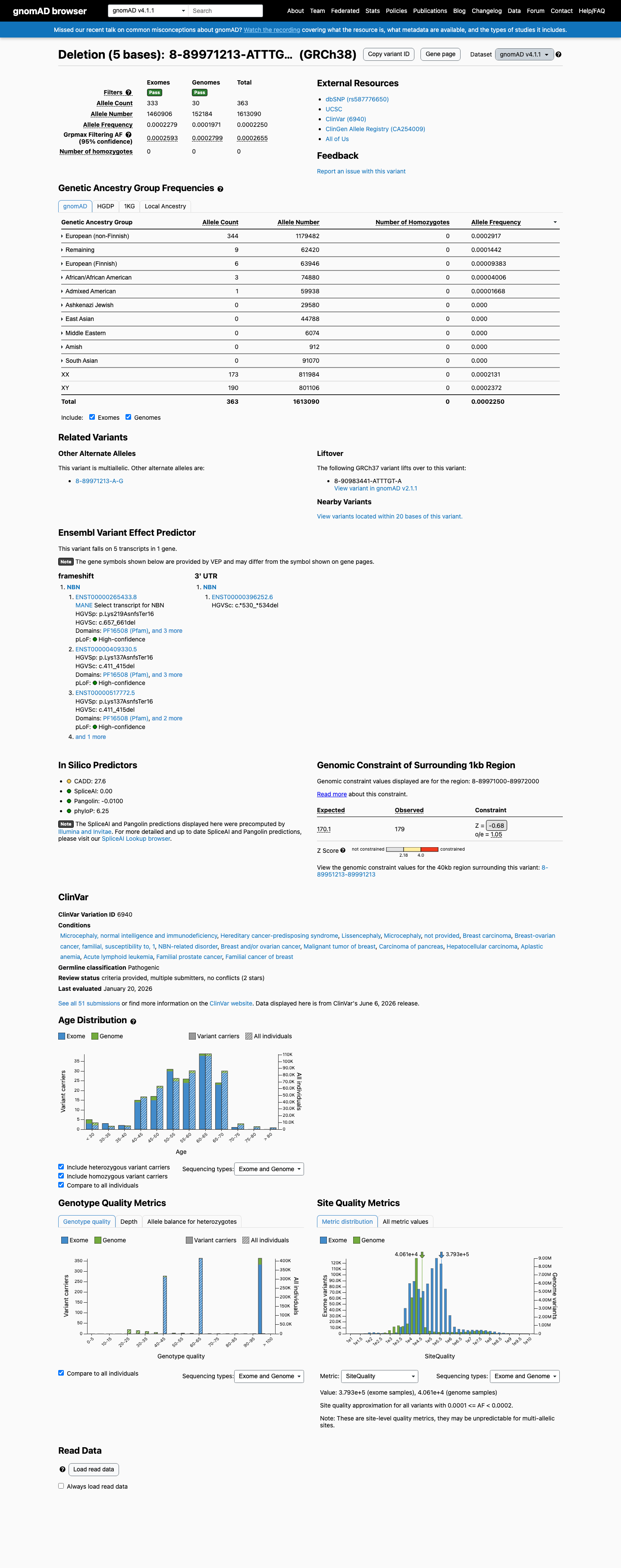
gnomAD v4.1
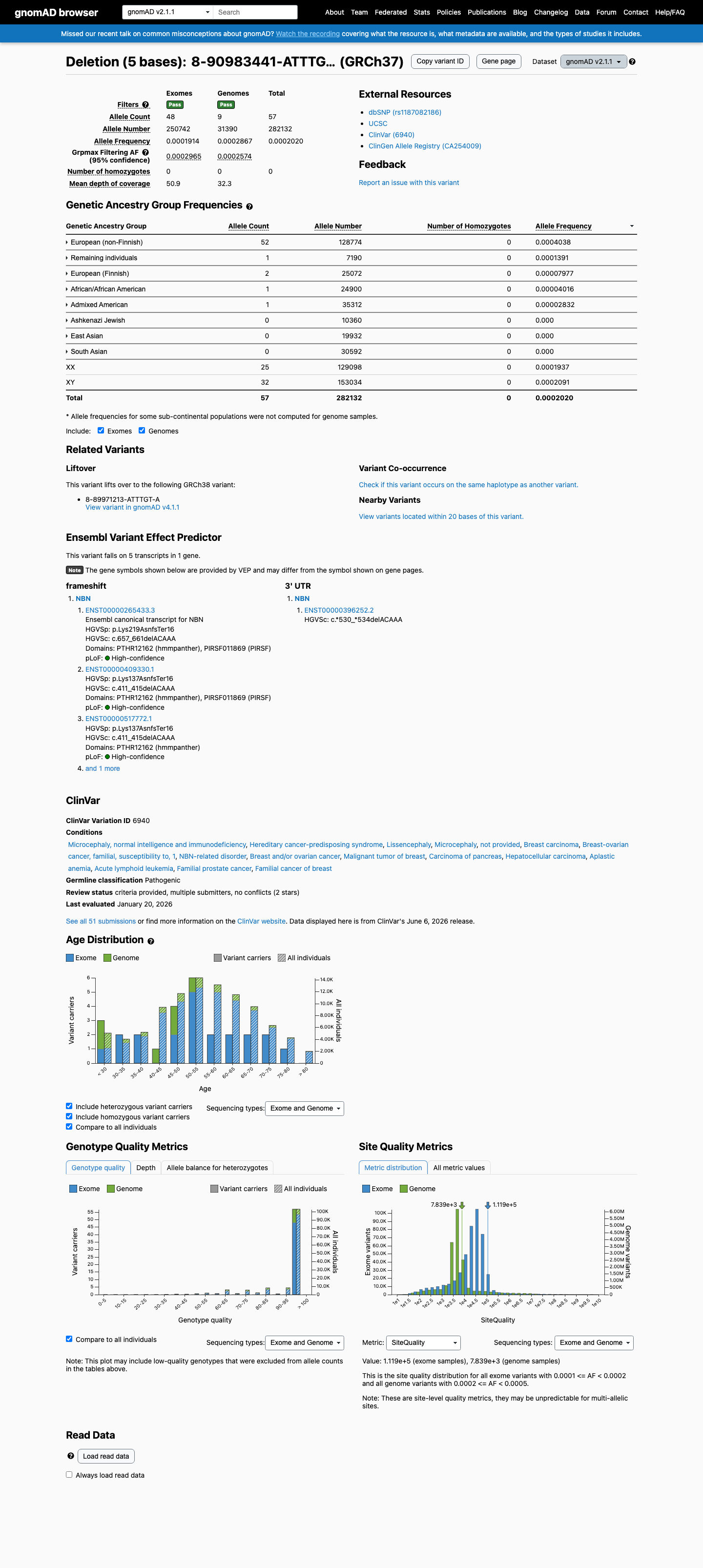
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000225034; MAF= 0.02250%, 363/1613090 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 0.000291653; MAF= 0.02917%, 344/1179482 alleles, homozygotes = 0); grpmax FAF= 0.00026547.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.000202033; MAF= 0.02020%, 57/282132 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 0.000403808; MAF= 0.04038%, 52/128774 alleles, homozygotes = 0); grpmax FAF= 0.0002965.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0003256975355553143, 6/18422 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.023%
· 363 / 1,613,090
0 hom · FAF 0.027%
0 hom · FAF 0.027%
European (non-Finnish) 344 / 1,179,482 |
0.029% |
Remaining individuals 9 / 62,420 |
0.014% |
European (Finnish) 6 / 63,946 |
0.0094% |
African/African American 3 / 74,880 |
0.004% |
Admixed American 1 / 59,938 |
0.0017% |
+ 5 not observed (Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish)
gnomAD v2.1
0.02%
· 57 / 282,132
0 hom · FAF 0.03%
0 hom · FAF 0.03%
European (non-Finnish) 52 / 128,774 |
0.04% |
Remaining individuals 1 / 7,190 |
0.014% |
European (Finnish) 2 / 25,072 |
0.008% |
African/African American 1 / 24,900 |
0.004% |
Admixed American 1 / 35,312 |
0.0028% |
+ 3 not observed (Ashkenazi Jewish, East Asian, South Asian)
gnomAD Canada 🇨🇦
0.033%
· 6 / 18,422
0 hom · FAF 0.022%
0 hom · FAF 0.022%
European (non-Finnish) 6 / 11,742 |
0.051% |
+ 8 not observed (African/African American, Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, Remaining individuals, South Asian)

ClinVar
This variant has been reported in ClinVar as Pathogenic (38 clinical laboratories) and as risk factor (1 clinical laboratory). (ClinVarID = 6940)
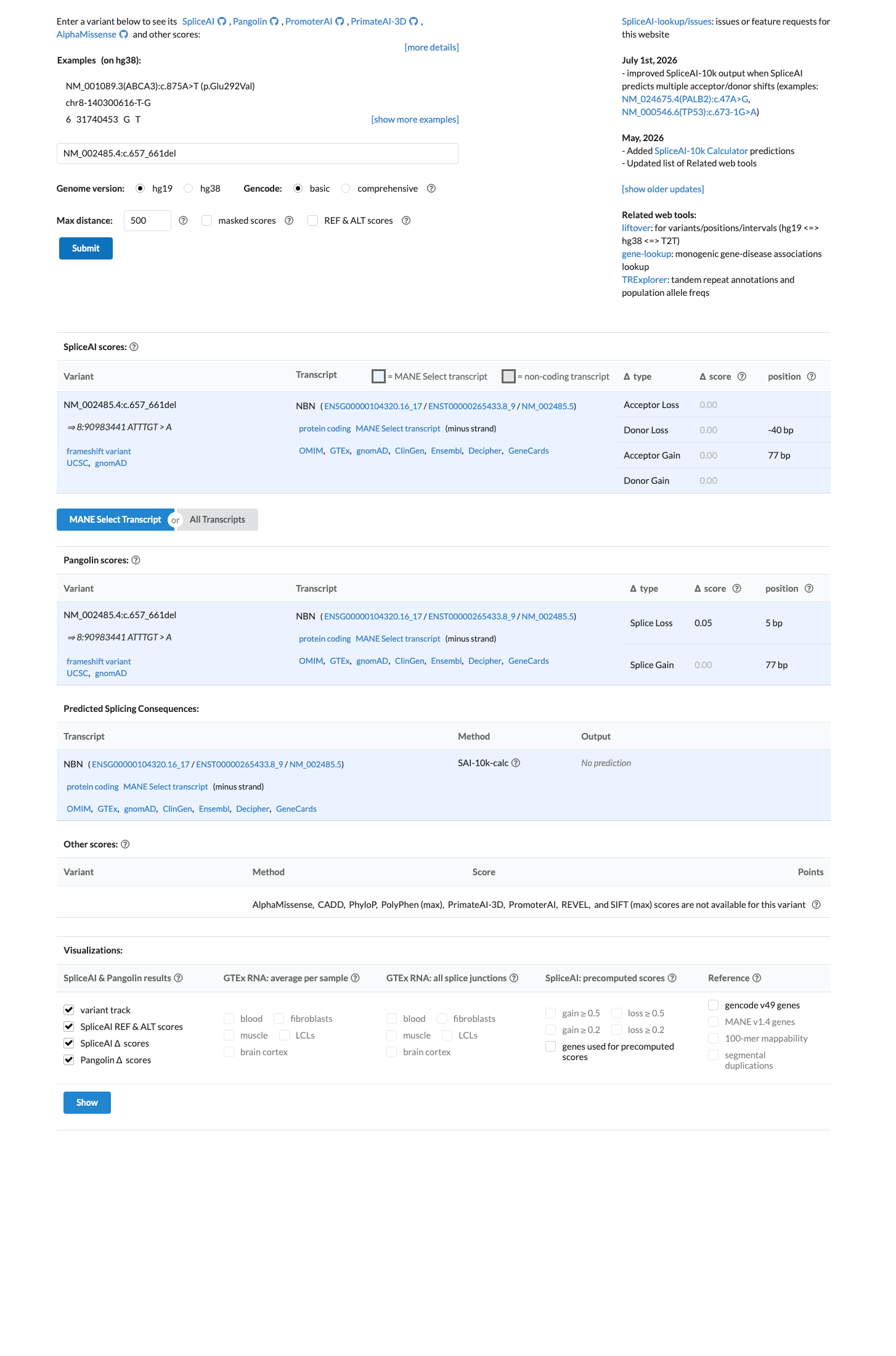
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
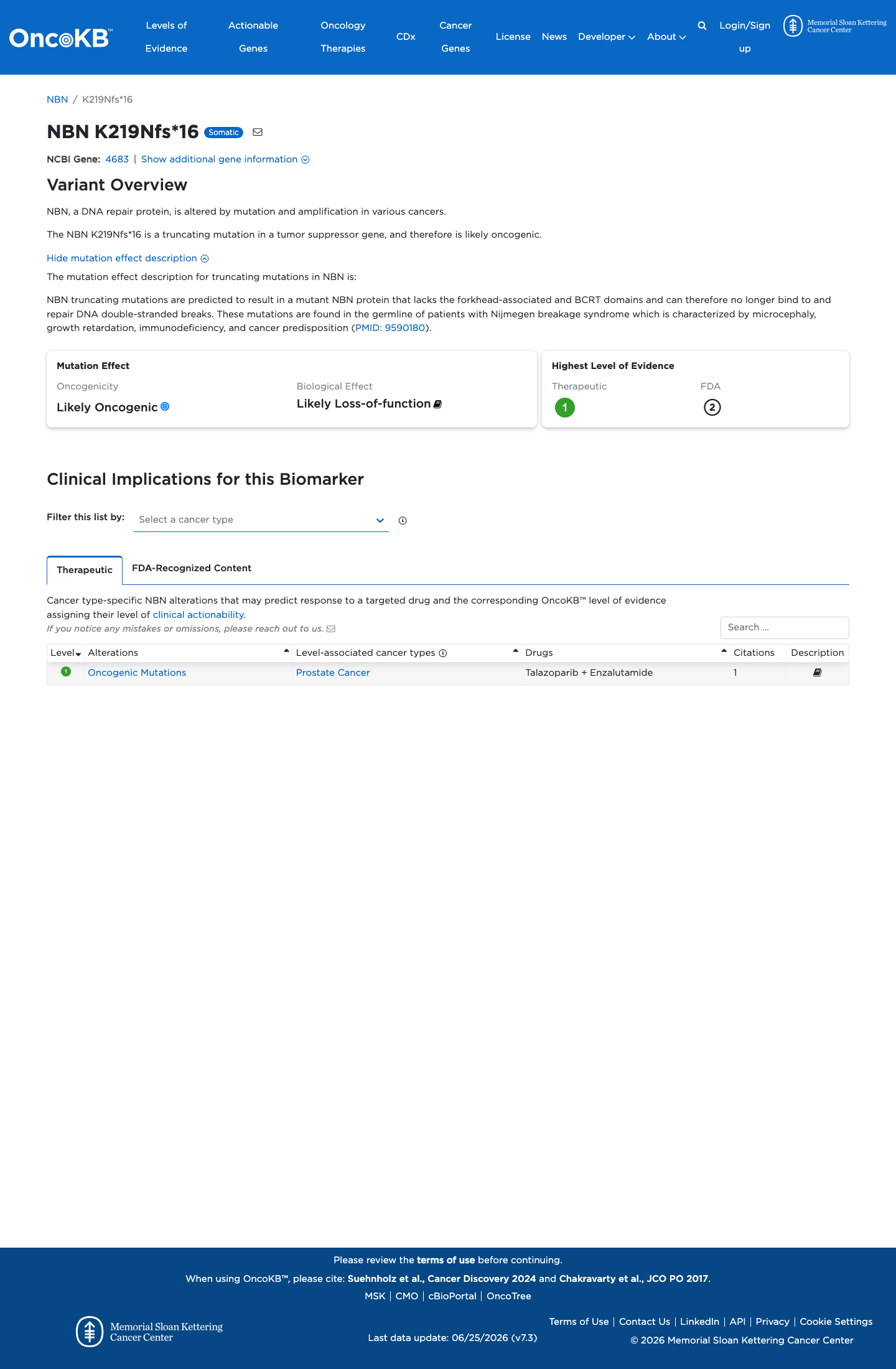
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV55370851, n = 6 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
8papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why.
Nijmegen breakage syndrome. The International Nijmegen Breakage Syndrome Study Group.
Searched
c.657_661del657del5657-661 del ACAAA
Found
The International Nijmegen Breakage Syndrome Study Group evaluated 55 NBS patients from the NBS registry. The majority carried the homozygous 657del5 (657-661delACAAA) truncating mutation in exon 6 of NBS1. Four additional truncating mutations were identified in patients with other haplotypes. No specific genotype-phenotype correlation was found; 40% of patients developed malignancy before age 21.
Variant
✓ Names this variant — characterised directly
Applied to
→PP1 supports · met
→PS4 supports · met
→PVS1 supports · met
Why
Variant confirmed as the major founder mutation in NBS patients; referenced in PVS1, PS4, and PP1 assessments.
Most of them had shown a truncating 5 bp deletion 657–661 delACAAA.
Location Abstract; Results, Table 1 (patient genotypes); Discussion · full text
Clinical presentation and mutation identification in the NBS1 gene in a boy with Nijmegen breakage syndrome.
Searched
c.657_661del657del5657-661
Found
Kleier et al. reported a 5-year-old Bosnian boy with severe microcephaly and chromosomal rearrangements involving chromosomes 7 and 14, initially diagnosed as ataxia telangiectasia. DNA analysis revealed homozygosity for the 657del5 mutation in NBS1 exon 6, confirming the diagnosis of Nijmegen breakage syndrome.
Variant
✓ Names this variant — characterised directly
Applied to
→PS4 supports · met
→PVS1 supports · met
Why
Variant confirmed homozygous in an NBS patient; supports PVS1 and PS4.
This patient is homozygous for the 657del5 mutation.
Location Abstract; Clinical Report; Discussion · full text
Clinical ascertainment of Nijmegen breakage syndrome (NBS) and prevalence of the major mutation, 657del5, in three Slav populations.
Searched
c.657_661del657del5657-661
Found
Varon et al. estimated the prevalence of the 657del5 founder mutation in three Slav populations by screening 4416 Guthrie cards. The carrier frequency was 1/177 overall, with the highest in Czech Republic (1/154). 82 of 84 unrelated NBS patients analyzed were homozygous for 657del5. NBS patients are often diagnosed late, with 14-37% already having malignancy at diagnosis.
Variant
✓ Names this variant — characterised directly
Applied to
→PS4 supports · met
→PVS1 supports · met
Why
Established high carrier frequency in Slav populations and near-complete homozygosity in NBS patients; supports PVS1 and PS4.
Most of the NBS patients known so far are of Slav origin and carry a major founder mutation 657del5 in exon 6 of the NBS1 gene.
Location Abstract; Results, Tables 1-2; Discussion · full text
An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele.
Searched
c.657_661del657del5K219Nfsp26p70
Found
Maser et al. demonstrated that the common NBS1 657del5 allele produces two protein products: a 26-kD N-terminal fragment (NBS1p26) containing FHA/BRCT domains but lacking the MRE11 interaction domain, and a 70-kD protein (NBS1p70) generated by internal translation initiation from cryptic ATG codons brought into frame by the deletion. NBS1p70 retains the MRE11 interaction domain and partially restores nuclear localization of the MRE11 complex but does not confer full function: NBS 657del5 LCLs exhibit radioresistant DNA synthesis. NBS1p70 is detected in lymphoblastoid cells but not fibroblasts.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
→PS3 supports · met
→PVS1 supports · met
Why
Direct variant-specific functional characterization; primary evidence for PS3 (moderate) and PM1. Evidence symmetry applied: same data evaluated for BS3 (not met) and PVS1 (does not negate).
NBS1p70 does not confer full function within the MRE11 complex.
Location Abstract; Results para 1-8; Figures 1-4 · Context EBV-transformed lymphoblastoid cell lines, untransformed fibroblasts; immunoprecipitation, western blot, in vitro transcription-translation, bicistronic expression constructs · full text
NBS1 is a prostate cancer susceptibility gene.
Searched
c.657_661del657del5657-661
Found
Cybulski et al. studied the 657del5 NBS1 founder allele in prostate cancer. The mutation was present in 5/56 (9%) familial prostate cancer cases (OR=16, P<0.0001), 7/305 (2.2%) nonfamilial cases (OR=3.9, P=0.01), and 9/1500 (0.6%) controls. LOH analysis showed loss of the wild-type NBS1 allele in 7/8 prostate tumors from mutation carriers versus 1/9 from noncarriers (P=0.003). The mutation segregated with prostate cancer in all 4 tested families.
Variant
✓ Names this variant — characterised directly
Applied to
→PP1 supports · met
→PS4 supports · met
Why
Strong case-control evidence for cancer risk; LOH data demonstrates functional hemizygosity in tumors. Referenced in PS4 (strong) and PP1 (moderate).
The NBS1 founder mutation was present in 5 of 56 (9%) patients with familial prostate cancer (odds ratio, 16; P < 0.0001).
Location Abstract; Results and Discussion; Figures 1-3 · full text
The importance of making ends meet: mutations in genes and altered expression of proteins of the MRN complex and cancer.
Searched
c.657_661del657del5657-661
Found
Dzikiewicz-Krawczyk reviewed the role of MRN complex genes in cancer. The 657del5 mutation is discussed as the most common NBS1 mutation, associated primarily with leukemia/lymphoma and breast cancer in heterozygous carriers. The review summarizes that the 657del5 allele produces both a truncated p26 (FHA/BRCT domains) and p70 fragment (MRE11 interaction domain), with p70 levels correlated with cancer occurrence in NBS patients.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
Why
Review article confirming the variant's association with multiple cancers and describing functional domain architecture; supports PM1.
Heterozygous NBS1 mutations and molecular variants 657del5, I171V, R215W and E185Q were most commonly analyzed. Among these, an association with cancer was found most frequently for 657del5.
Location Section 3, Table 1; Section 2 · full text
Heterozygous germ-line mutations in the NBN gene predispose to medulloblastoma in pediatric patients.
Searched
c.657_661del657del5657-661
Found
Ciara et al. screened 104 Polish pediatric medulloblastoma patients for NBN mutations and identified 7 heterozygous carriers (6.7%). Three carried c.657_661del5 and four carried c.511A>G. The c.657_661del5 carrier frequency in medulloblastoma was 2.88% versus 0.59% in population controls (OR=4.86, P=0.0028). For classic-type medulloblastoma, the OR was 6.83 (P=0.0001). No LOH was observed in the 5 tested tumors from NBN mutation carriers.
Variant
✓ Names this variant — characterised directly
Applied to
→PP1 supports · met
→PS4 supports · met
Why
Case-control study demonstrating increased medulloblastoma risk in 657del5 carriers; referenced in PS4 and PP1.
heterozygous carriers of the germ-line NBN gene mutations (c.511A>G and c.657_661del5) may exhibit increased susceptibility to developing MB.
Location Abstract; Results, Tables 1-4; Discussion · full text
Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome.
Searched
c.657_661del657del5657-661
Found
Varon et al. positionally cloned the NBN (NBS1) gene and identified nibrin as a novel DNA double-strand break repair protein. The 657del5 mutation was identified as the major mutation in NBS patients. This is the seminal discovery paper for the NBN gene and the 657del5 founder mutation.
Variant
✓ Names this variant
Applied to
→PVS1 supports · met
Why
Seminal discovery paper for NBN and 657del5; supports gene-disease relationship and PVS1. Full text not available in case folder.
We describe the positional cloning of a gene encoding a novel protein, nibrin, is mutated in Nijmegen breakage syndrome.
Location Abstract (snippet); full text not available
Sources & reference links