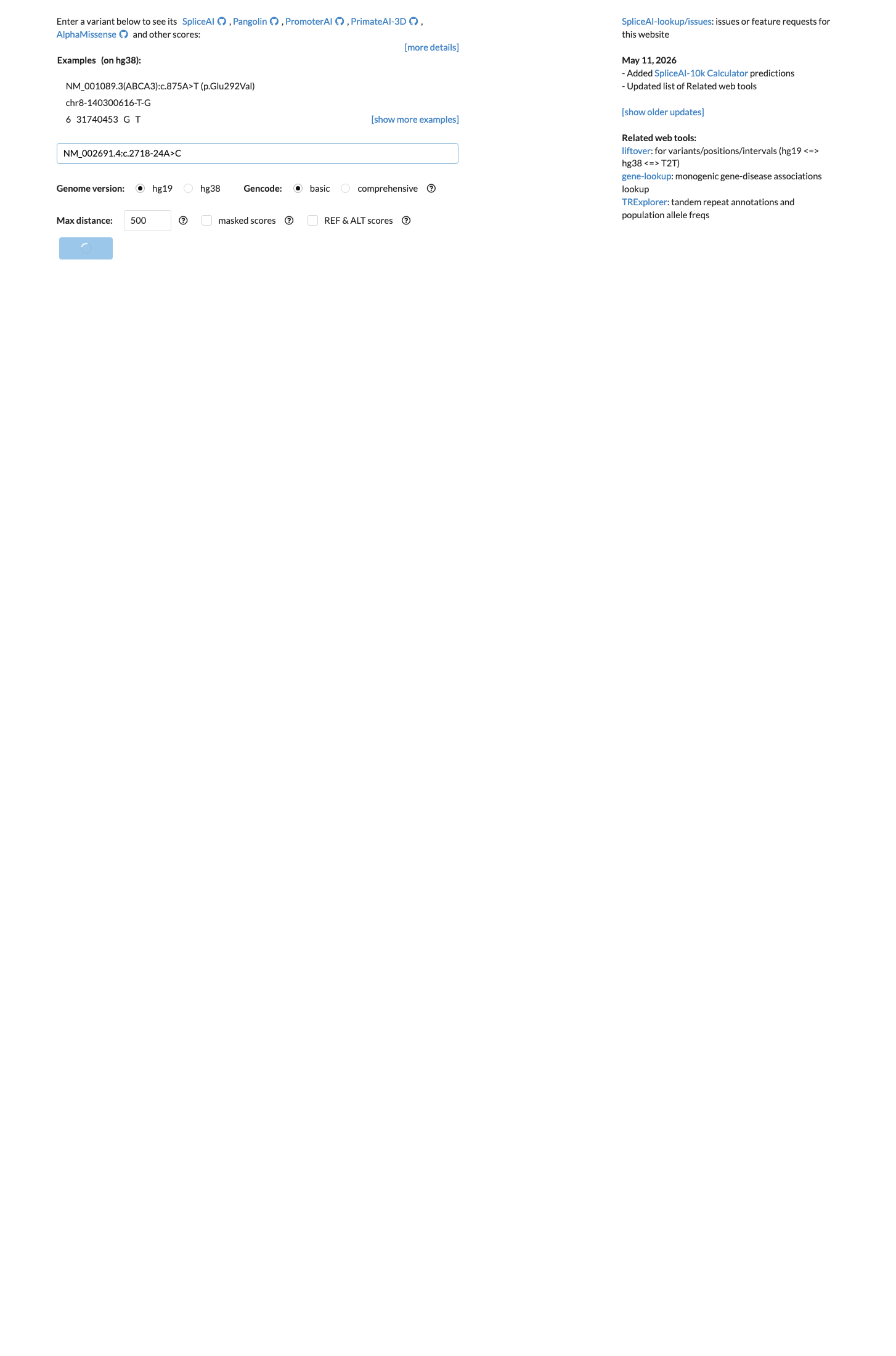
NM_002691.4:c.2718-24A>C is an intronic variant in POLD1 located 24 bases upstream of exon 21, outside the canonical splice consensus region. SpliceAI predicts no significant splicing impact (max delta score = 0.00), indicating this variant is unlikely to alter normal splicing (BP4).1 The variant is present in gnomAD v2.1 at an allele frequency of 0.458% (358/78,106 alleles) and in v4.1 at 0.494% (2,810/568,564 alleles), far exceeding the maximum credible population frequency for a highly penetrant rare dominant disorder such as polymerase proofreading-associated polyposis (BS1).2 The variant is absent from ClinVar with no disease associations or submissions, and no publications were identified that specifically mention this variant.3 Based on the generic ACMG/AMP 2015 classification framework (PMID:25741868), the combination of one strong benign criterion (BS1) and one supporting benign criterion (BP4) supports a classification of Likely Benign.4
POLD1
Final classification
Likely Benign
POLD1 c.2718-24A>C · p.?
POLD1
NM_002691.4:c.2718-24A>C is an intronic variant in POLD1 located 24 bases upstream of exon 21, outside the canonical splice consensus region.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS1 strong benign, BP4 supporting benign; combination = 1 strong benign + 1 supporting benign, which maps to Likely Benign.
Classification rationale
BS1BP4
Likely Benign
POLD1 c.2718-24A>C
BS1 + BP4
→
Likely Benign
Gene diagram
· NM_002691.4 · variants mapped to exon structure
POLD1
NM_002691.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 17 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
strong
Benign
This variant is present at a frequency that greatly exceeds the maximum expected for a highly penetrant rare dominant disorder: gnomAD v2.1 allele frequency is 0.458% (358/78,106 alleles) and v4.1 is 0.494% (2,810/568,564 alleles), both above the BS1 threshold of >0.3%. The grpmax filtering allele frequency is 6.53% (v2.1) and 2.04% (v4.1), further supporting that this is a common polymorphism.
gnomAD v2.1 AF=0.458% (>0.3%)grpmax FAF=6.53%gnomAD v4.1 AF=0.494% (>0.3%)
✓
BP4
supporting
Benign
Multiple lines of computational evidence suggest no deleterious impact: SpliceAI predicts no significant splicing effect (max delta score = 0.00), with no evidence of cryptic splice site creation or disruption. REVEL and BayesDel are not applicable to this intronic variant but their absence does not contradict the benign assessment.
SpliceAI max delta score = 0.00no predicted impact on splicingno computational evidence of pathogenicity.
Assessed · not applied
Pathogenic
PS2
No de novo occurrence report with confirmed maternity and paternity was identified for this variant in the literature; a de novo event must be directly observed in a published study to apply PS2.
PS3
No well-established in vitro or in vivo functional studies supporting a damaging effect on the gene product were identified for this specific variant.
PS4
No case-control data or enriched observation in affected individuals beyond population frequency is available; the variant is absent from ClinVar with no curated disease associations.
PM1
This intronic variant is not located within a well-established functional domain or mutational hot spot; cancer hotspot analysis confirms no residue-level significance.
PM2
This variant is present in gnomAD at an allele frequency of 0.458% (v2.1, 358/78,106 alleles) and 0.494% (v4.1, 2,810/568,564 alleles), both well above the PM2 threshold of <0.1%.
PM6
No de novo observation (without confirmation of maternity/paternity) was identified for this variant; a de novo report must be directly read from a published source to apply PM6.
PP1
No cosegregation data in affected family members is available for this variant.
PP3
Multiple lines of computational evidence do not support a deleterious effect: SpliceAI predicts no significant splicing impact (max delta = 0.00); REVEL and BayesDel are not applicable to this intronic variant.
PP4
No patient phenotype or family history information is available for this variant; PP4 requires a phenotype highly specific for a disease with a single genetic etiology.
PP5
No reputable source has reported this variant as pathogenic; the variant is absent from ClinVar with zero submissions.
Benign
BA1
The overall allele frequency in gnomAD is 0.458% (v2.1) and 0.494% (v4.1), both below the BA1 threshold of >1% for a dominant disorder.
BS2
Although the variant is observed in many heterozygous carriers across gnomAD (358 alleles in v2.1, 2,810 in v4.1), gnomAD includes individuals ascertained through disease studies and does not constitute a confirmed healthy adult control cohort; BS2 requires observation in healthy adults with full expected penetrance.
BS3
No well-established in vitro or in vivo functional studies demonstrating no damaging effect on the gene product were identified for this specific variant.
BS4
No family cosegregation data demonstrating lack of segregation with disease is available for this variant.
BP2
No data available regarding observation in trans with a pathogenic variant for a fully penetrant dominant gene, or in cis with a pathogenic variant in any inheritance pattern.
BP5
No case has been identified in which this variant is found in a patient with an alternate molecular basis for disease.
BP6
No reputable source has reported this variant as benign; the variant is absent from ClinVar with zero submissions.
N/A · 9
PVS1 · PS1 · PM3 · PM4 · PM5 · PP2 · BP1 · BP3 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
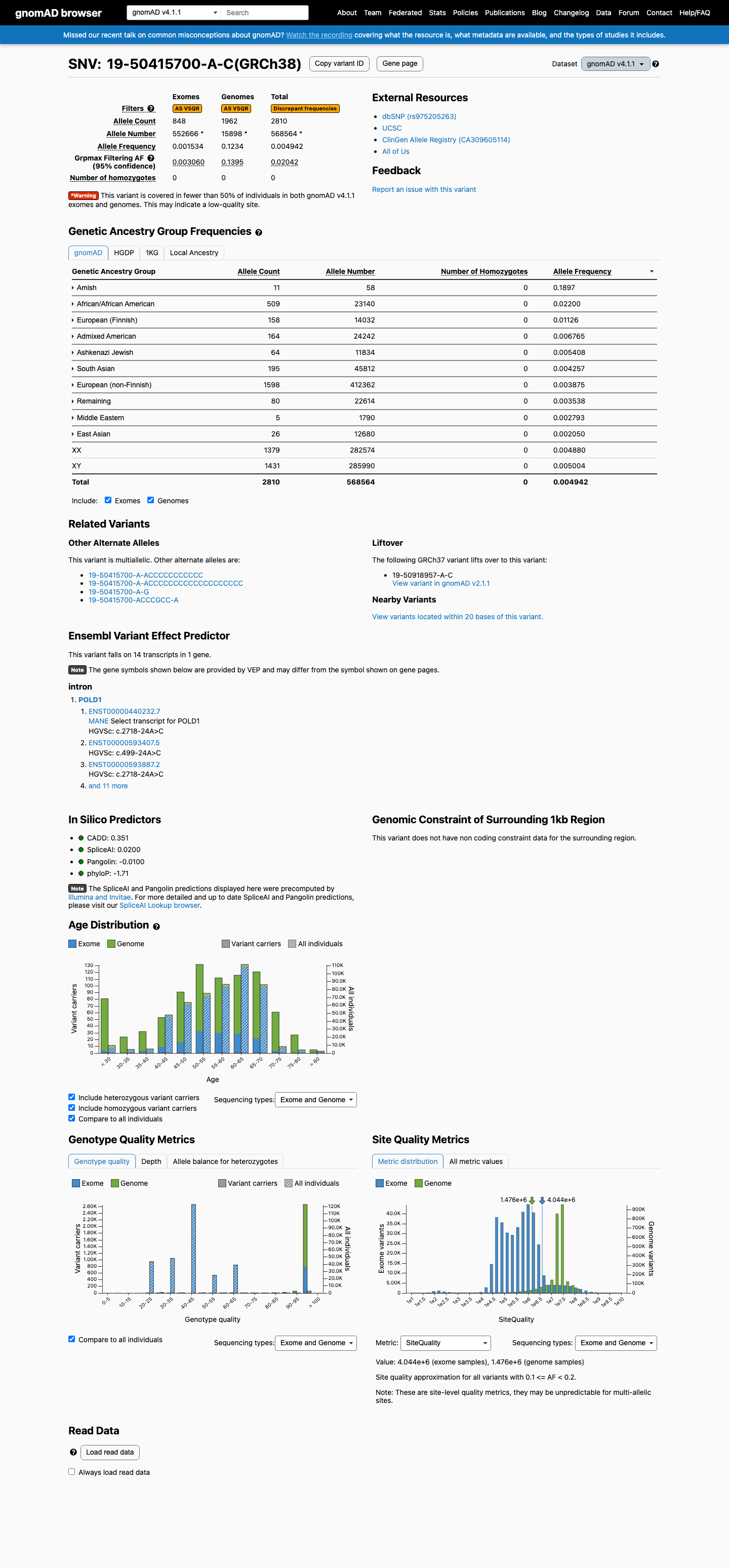
This variant is present in gnomAD v4.1 (AF= 0.00494228; MAF= 0.49423%, 2810/568564 alleles, homozygotes = 0) and has highest observed frequency in the Amish population (AF= 0.189655; MAF= 18.96552%, 11/58 alleles, homozygotes = 0); grpmax FAF= 0.0204173.
v2.1
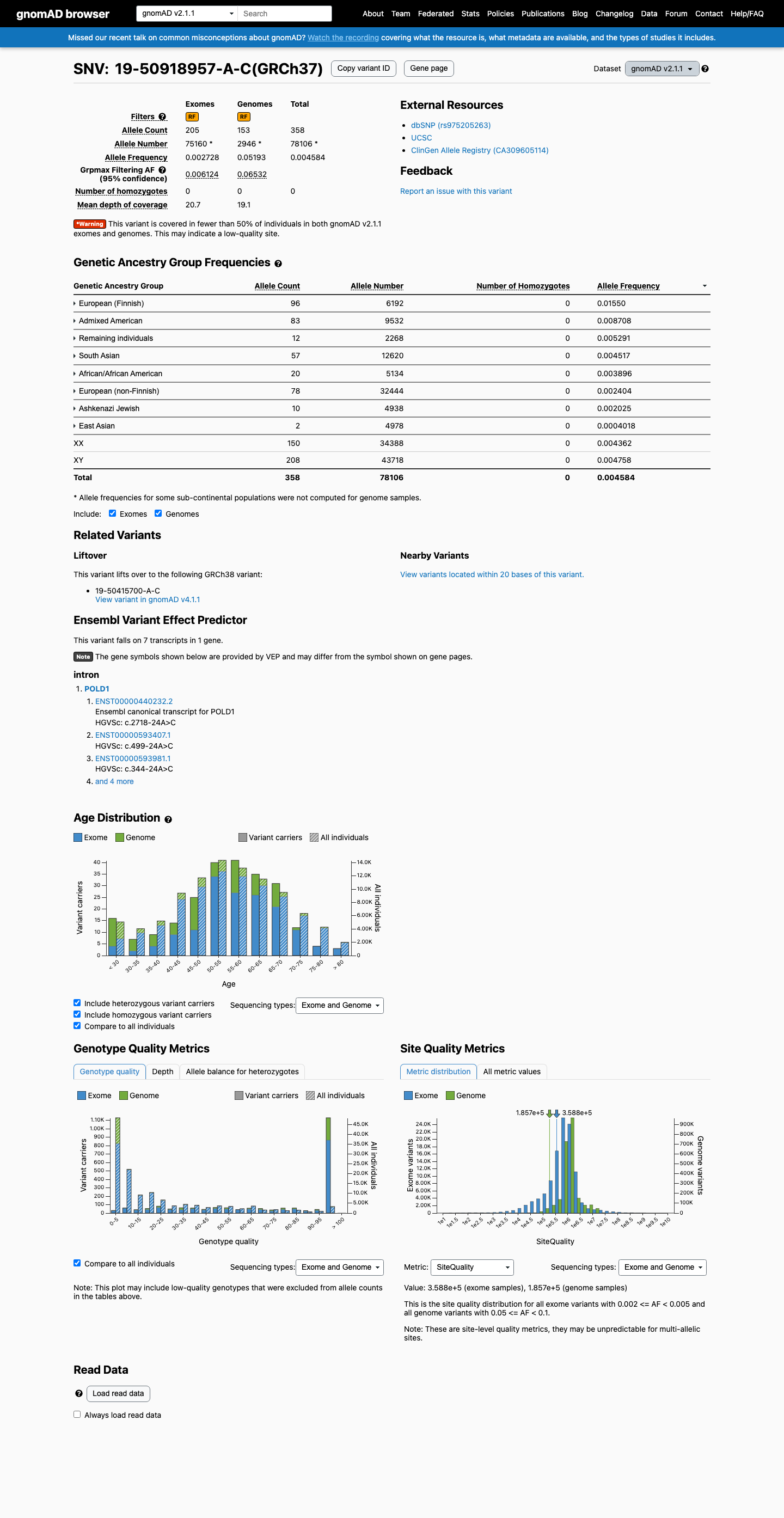
This variant is present in gnomAD v2.1 (AF= 0.00458351; MAF= 0.45835%, 358/78106 alleles, homozygotes = 0) and has highest observed frequency in the European (Finnish) population (AF= 0.0155039; MAF= 1.55039%, 96/6192 alleles, homozygotes = 0); grpmax FAF= 0.0653219.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.49%
· 2810 / 568,564
0 hom · FAF 2%
0 hom · FAF 2%
Amish 11 / 58 |
19% |
African/African American 509 / 23,140 |
2.2% |
European (Finnish) 158 / 14,032 |
1.1% |
Admixed American 164 / 24,242 |
0.68% |
Ashkenazi Jewish 64 / 11,834 |
0.54% |
South Asian 195 / 45,812 |
0.43% |
European (non-Finnish) 1598 / 412,362 |
0.39% |
Remaining individuals 80 / 22,614 |
0.35% |
Middle Eastern 5 / 1,790 |
0.28% |
East Asian 26 / 12,680 |
0.21% |
gnomAD v2.1
0.46%
· 358 / 78,106
0 hom · FAF 6.5%
0 hom · FAF 6.5%
European (Finnish) 96 / 6,192 |
1.6% |
Admixed American 83 / 9,532 |
0.87% |
Remaining individuals 12 / 2,268 |
0.53% |
South Asian 57 / 12,620 |
0.45% |
African/African American 20 / 5,134 |
0.39% |
European (non-Finnish) 78 / 32,444 |
0.24% |
Ashkenazi Jewish 10 / 4,938 |
0.2% |
East Asian 2 / 4,978 |
0.04% |
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links