NM_002878.4:c.198G>T (p.Val66=) is a synonymous variant in RAD51D with no predicted amino acid change.1 SpliceAI predicts no splice impact for this variant (max delta score = 0.00), meeting BP7 (supporting).2 This variant is present in gnomAD v4.1 with a grpmax filtering allele frequency of 0.303% and an East Asian subpopulation frequency of 0.348% (156/44,880 alleles, one homozygote), marginally exceeding the 0.3% threshold for BS1 at supporting strength.3 Multiple clinical diagnostic laboratories have classified this variant as Likely benign (5 labs) or Benign (1 lab) in ClinVar (Variation ID: 184496), consistent with BP6 at supporting strength.4 No variant-specific functional, segregation, de novo, or case-control data were identified in the literature. Three PMIDs from ClinVar (25741868, 25394175, 28492532) are guideline or methods papers that do not mention NM_002878.4:c.198G>T.
RAD51D
Final classification
Likely Benign
RAD51D c.198G>T · p.Val66=
RAD51D
NM_002878.4:c.198G>T (p.Val66=) is a synonymous variant in RAD51D with no predicted amino acid change.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS1 supporting, BP6 supporting, BP7 supporting; combination = 3 supporting benign, which maps to Likely Benign.
Classification rationale
BS1BP6BP7
Likely Benign
RAD51D c.198G>T
BS1 + BP6 + BP7
→
Likely Benign
Gene diagram
· NM_002878.4 · variants mapped to exon structure
RAD51D
NM_002878.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 15 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
supporting
Benign
The gnomAD v4.1 grpmax filtering allele frequency is 0.303%, marginally exceeding the 0.3% threshold for BS1. In the East Asian subpopulation, the allele frequency is 0.348% (156/44,880 alleles) with one homozygote observed, indicating the variant is too common in the general population to be a highly penetrant pathogenic variant for RAD51D-associated autosomal dominant cancer predisposition. The borderline frequency and the v2.1 grpmax FAF of 0.022% (below threshold) warrant supporting rather than strong strength.
gnomAD v4.1 grpmax FAF = 0.303% (>0.3% BS1 threshold)gnomAD v4.1 East Asian AF = 0.348% (156/44880) with 1 homozygote
✓
BP6
supporting
Benign
Multiple reputable clinical diagnostic laboratories have classified NM_002878.4:c.198G>T as Likely benign (Labcorp/Invitae, Color Health, GeneDx, Ambry Genetics, Counsyl) or Benign (Myriad Genetics) in ClinVar (Variation ID: 184496). The underlying evidence criteria are not publicly available for independent evaluation, so BP6 is applied at supporting strength.
ClinVar Likely benign: Labcorp/Invitae (SCV000561575)Color Health (SCV000537457)GeneDx (SCV000514355)
✓
BP7
supporting
Benign
NM_002878.4:c.198G>T is a synonymous variant (p.Val66=) with no predicted amino acid change. SpliceAI predicts no splice impact (max delta score = 0.00), and no novel splice site creation is predicted. This meets the ACMG/AMP BP7 criterion for synonymous variants without predicted splicing consequences.
Synonymous variant p.Val66= (no amino acid change)SpliceAI max delta = 0.00 (no predicted splice impactno novel splice site)
Assessed · not applied
Pathogenic
PS2
No de novo data available for NM_002878.4:c.198G>T.
PS3
No well-established functional studies identified for this specific variant.
PS4
The variant is present in gnomAD at low frequency (v2.1: 0.004%, v4.1: 0.010%) with one homozygote in v4.1.
PM1
This variant does not lie in a statistically significant mutational hotspot in RAD51D.
PM2
While the overall gnomAD allele frequency is below the 0.1% PM2 threshold (v2.1: 0.004%, v4.1: 0.010%), the gnomAD v4.1 grpmax filtering allele frequency is 0.303%, meeting BS1.
PM6
No de novo data available for NM_002878.4:c.198G>T.
PP1
No segregation data available for NM_002878.4:c.198G>T.
PP4
No phenotype or disease-specificity data available for NM_002878.4:c.198G>T.
PP5
PP5 requires a reputable source to report the variant as pathogenic.
Benign
BA1
The gnomAD grpmax filtering allele frequency is 0.303% (v4.1), which is well below the 1% BA1 threshold.
BS2
While one homozygote is observed in gnomAD v4.1, the individuals in gnomAD are not specifically confirmed as healthy adults.
BS3
No well-established functional studies showing no damaging effect were identified for NM_002878.4:c.198G>T.
BS4
No segregation data available for NM_002878.4:c.198G>T.
BP2
No data on observation of NM_002878.4:c.198G>T in trans with a known pathogenic RAD51D variant.
BP4
BP4 (multiple lines of computational evidence suggest no impact) is subsumed by BP7 for this synonymous variant.
N/A · 10
PVS1 · PS1 · PM3 · PM4 · PM5 · PP2 · PP3 · BP1 · BP3 · BP5
Research & evidence
Population frequency · supports benign
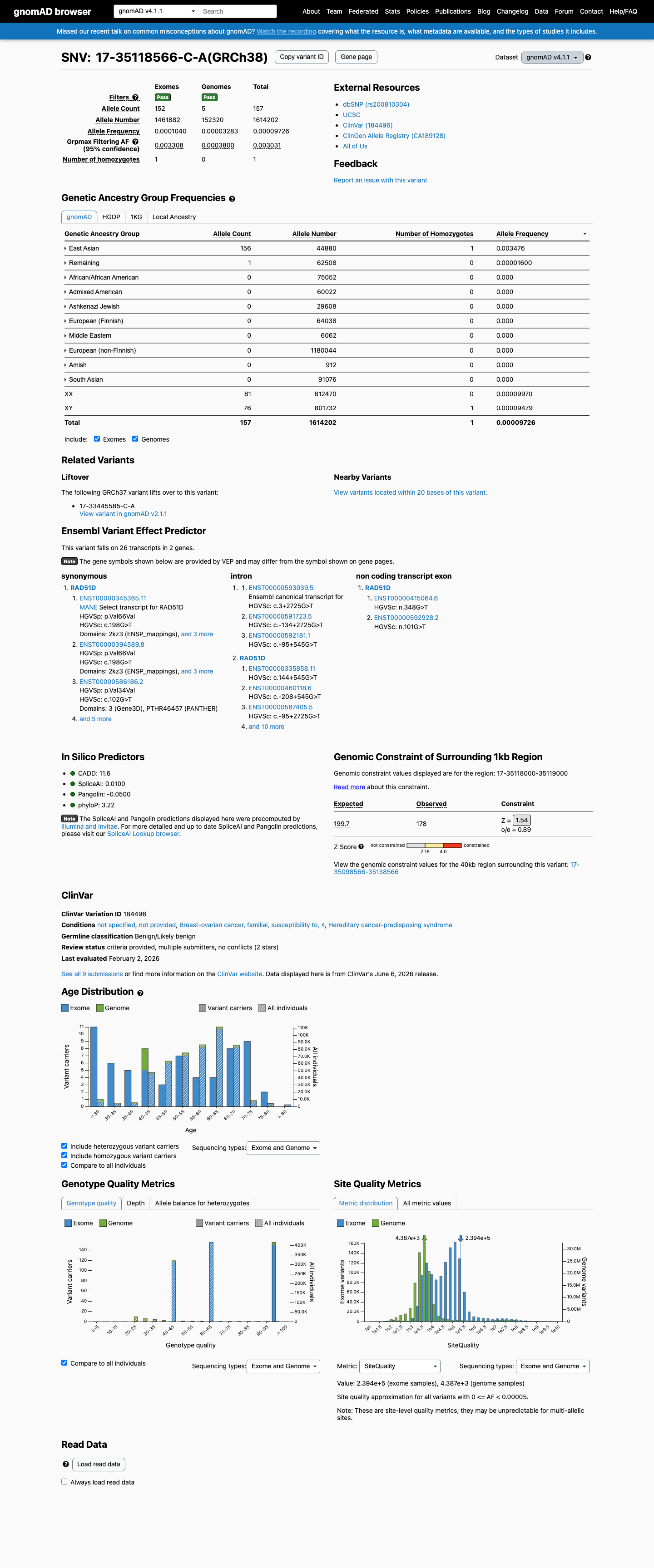
gnomAD v4.1
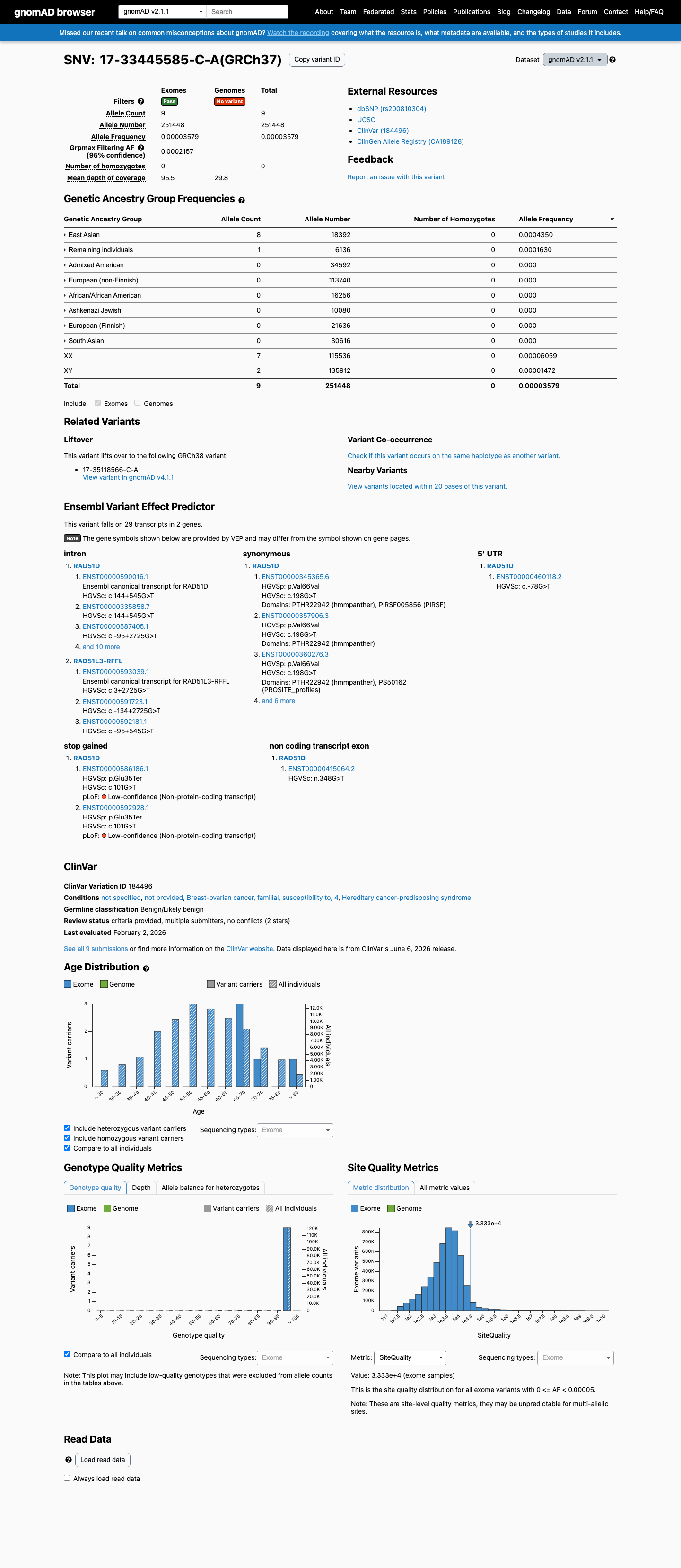
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 9.72617e-05; MAF= 0.00973%, 157/1614202 alleles, homozygotes = 1) and has highest observed frequency in the East Asian population (AF= 0.00347594; MAF= 0.34759%, 156/44880 alleles, homozygotes = 1); grpmax FAF= 0.00303071.
v2.1
This variant is present in gnomAD v2.1 (AF= 3.57927e-05; MAF= 0.00358%, 9/251448 alleles, homozygotes = 0) and has highest observed frequency in the East Asian population (AF= 0.000434972; MAF= 0.04350%, 8/18392 alleles, homozygotes = 0); grpmax FAF= 0.00021573.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0097%
· 157 / 1,614,202
1 hom · FAF 0.3%
1 hom · FAF 0.3%
East Asian 156 / 44,880 |
0.35% 1 hom |
Remaining individuals 1 / 62,508 |
0.0016% |
+ 8 not observed (Admixed American, European (Finnish), Amish, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American, European (non-Finnish))
gnomAD v2.1
0.0036%
· 9 / 251,448
0 hom · FAF 0.022%
0 hom · FAF 0.022%
East Asian 8 / 18,392 |
0.043% |
Remaining individuals 1 / 6,136 |
0.016% |
+ 6 not observed (African/African American, Admixed American, Ashkenazi Jewish, European (Finnish), European (non-Finnish), South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
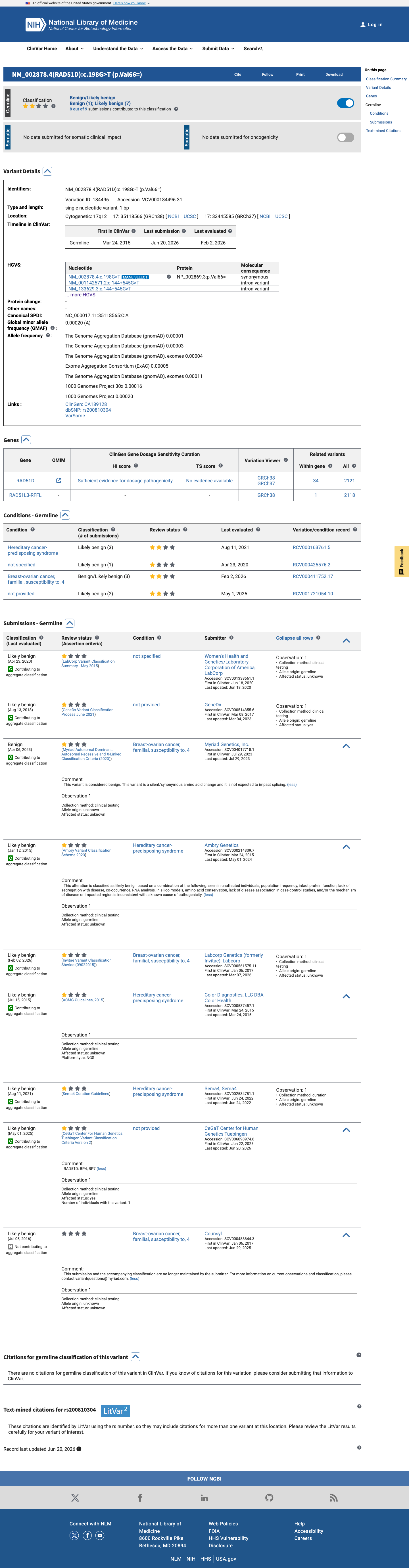
ClinVar
This variant has been reported in ClinVar as Likely benign (7 clinical laboratories) and as Benign (1 clinical laboratory). (ClinVarID = 184496)
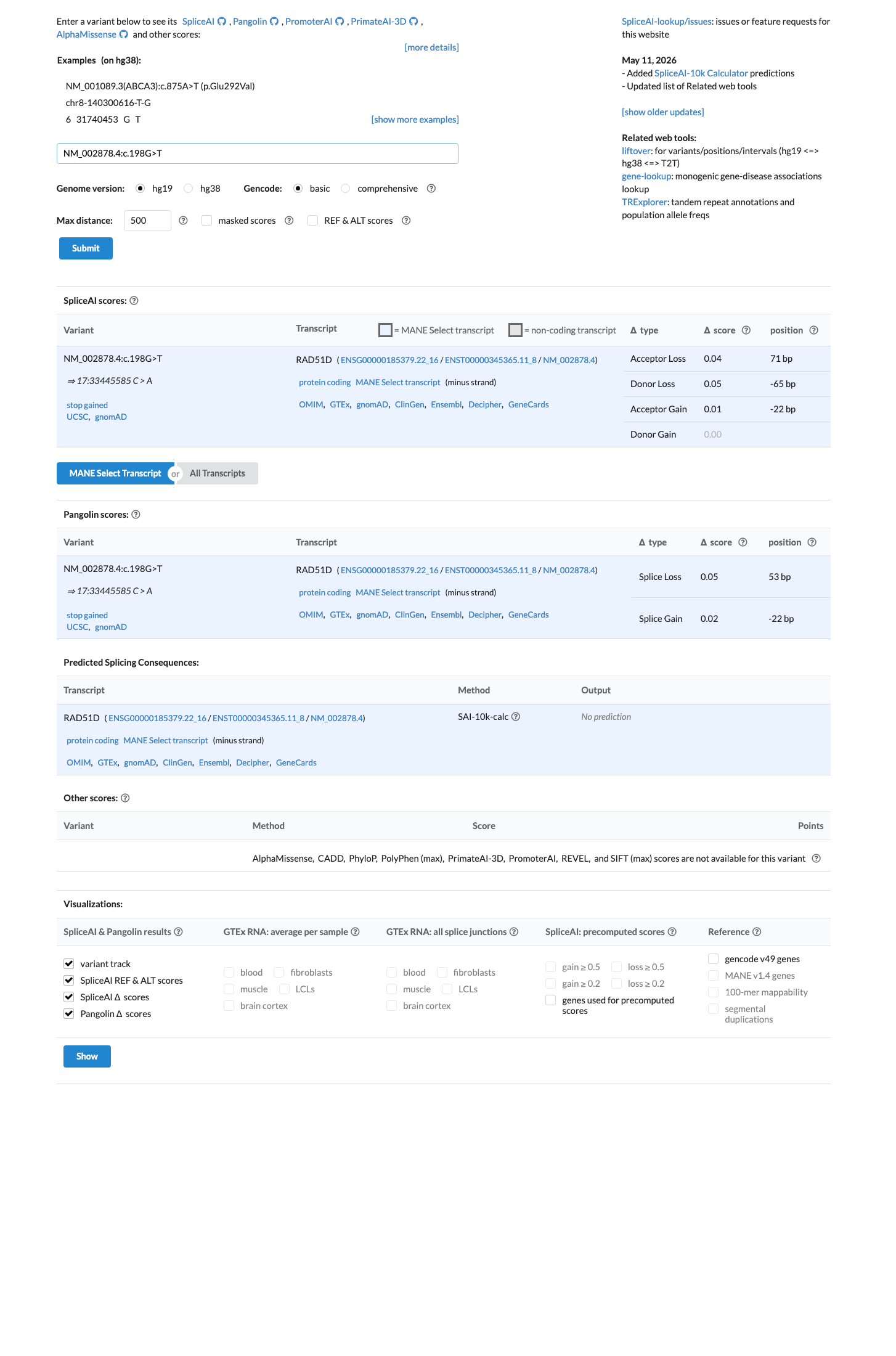
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 3 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR