MAP2K4 c.400C>T (p.Arg134Trp) is a missense variant located at a well-established mutational hotspot (PM1_moderate) in the β3-αC loop of the kinase domain. It is the most frequent MKK4 mutation catalogued in COSMIC and lies within a statistically significant hotspot domain as defined by cancerhotspots.org.1 This variant is absent from gnomAD v2.1 and gnomAD-Canada, and is observed at an extremely low frequency in gnomAD v4.1 (1/1,613,378 alleles; AF=6.2×10⁻⁷), meeting the PM2 criterion for very low population frequency (PM2_moderate).2 Computational in silico predictors are indeterminate: REVEL (0.475) and BayesDel (0.247) fall below established pathogenic thresholds, and SpliceAI predicts no splice alteration. PP3 is not met.3 Molecular dynamics simulations (PMID:33101607) demonstrate that p.Arg134Trp introduces a TRP134-TRP134 π-π interaction that distorts the N-lobe dimer interface of autoinhibited MKK4, but this computational evidence does not independently meet PS3 criteria for well-established functional evidence.4 The variant is absent from ClinVar and no clinical laboratory or expert panel classification is available. No de novo, segregation, case-control, or experimental functional data were identified for this variant.5 Two pathogenic criteria of moderate strength are met (PM1 and PM2). Under generic ACMG/AMP 2015 combination rules (PMID:25741868), two moderate criteria are insufficient to reach a Likely Pathogenic classification, which requires at least three moderate criteria or two moderate plus two supporting criteria. The variant is classified as a Variant of Uncertain Significance (VUS).6
MAP2K4
Final classification
VUS
MAP2K4 c.400C>T · p.Arg134Trp
MAP2K4
MAP2K4 c.400C>T (p.Arg134Trp) is a missense variant located at a well-established mutational hotspot (PM1_moderate) in the β3-αC loop of the kinase domain. It is the most frequent MKK4 mutation catalogued in COSMIC and lies within a statistically significant hotspot domain as defined by cancerhotspots.org.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM1 moderate, PM2 moderate; combination = 2 moderate, which maps to VUS.
Classification rationale
PM1PM2
VUS
MAP2K4 c.400C>T
PM1 + PM2
→
VUS
3
revelbayesdelspliceai ↗
6
generic_acmg_combination_rules
Gene diagram
· NM_003010.3 · variants mapped to exon structure
MAP2K4
NM_003010.3
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 19 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM1
moderate
Pathogenic
p.Arg134Trp (R134W) lies at a well-established mutational hotspot in MAP2K4. It is identified as one of two COSMIC hotspot mutations (R134W and S184L) and is located in a statistically significant hotspot domain (β3-αC loop), as confirmed by cancerhotspots.org and PMID:33101607.
Statistically significant hotspot at residue 134R134W is the most frequent MKK4 mutation in COSMIC (PMID:33101607)cancerhotspots.org also confirms hotspot significance at this residue
✓
PM2
moderate
Pathogenic
NM_003010.3:c.400C>T is absent from gnomAD v2.1 and gnomAD-Canada, and is present in gnomAD v4.1 at an extremely low allele frequency of 6.2×10⁻⁷ (1/1,613,378 alleles; highest subpopulation NFE AF=8.5×10⁻⁷). This frequency is well below the 0.1% threshold for PM2.
gnomAD v2.1: absentgnomAD v4.1: 1/1613
Assessed · not applied
Pathogenic
PS1
No prior observation of a different nucleotide change at the same amino acid (p.Arg134) with a pathogenic classification has been identified in ClinVar or published literature.
PS2
No de novo observation with confirmed maternity and paternity has been identified for NM_003010.3:c.400C>T.
PS3
No well-established in vitro or in vivo functional assay data exist for p.Arg134Trp.
PS4
No case-control or aggregate cohort data comparing variant prevalence in affected individuals versus controls have been identified for this variant.
PM6
No de novo observation (assumed de novo without confirmation of maternity/paternity) has been identified for NM_003010.3:c.400C>T.
PP1
No co-segregation data in affected family members have been identified for this variant.
PP2
Insufficient data to determine whether MAP2K4 has a low rate of benign missense variation and whether missense variants are a common mechanism of disease.
PP3
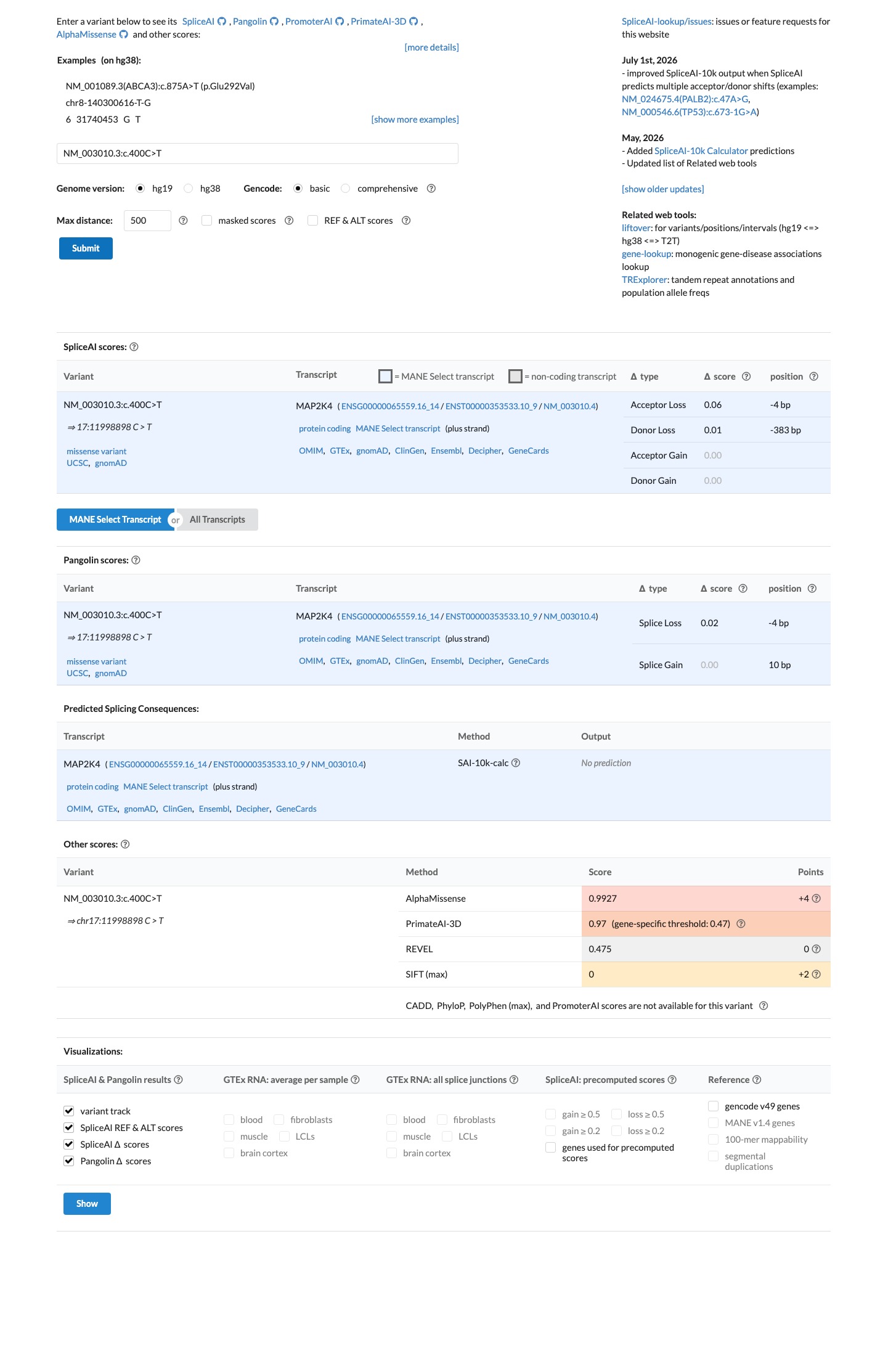
In silico predictors do not support a deleterious effect: REVEL score is 0.475 (below 0.5 pathogenic threshold), BayesDel score is 0.247 (below ~0.27 pathogenic threshold), and SpliceAI max delta score is 0.00 indicating no predicted splice impact.
PP4
No patient phenotype or family history data are available to assess whether the clinical presentation is highly specific for a MAP2K4-related disorder.
PP5
No reputable source (diagnostic laboratory, expert panel) has reported this variant as pathogenic.
Benign
BA1
The variant has a maximum population allele frequency of 8.5×10⁻⁷ in gnomAD v4.1 (NFE), far below the BA1 threshold of >1%.
BS1
The variant has a total allele frequency of 6.2×10⁻⁷ in gnomAD v4.1, far below the BS1 threshold of >0.3%.
BS2
No observation of this variant in healthy adult individuals in the context of a fully penetrant disorder expected to manifest at an early age has been identified.
BS3
No well-established in vitro or in vivo functional studies demonstrating no damaging effect have been identified.
BS4
No segregation data in affected families are available to demonstrate lack of co-segregation with disease.
BP1
BP1 applies when missense variants occur in a gene where primarily truncating variants cause disease.
BP4
Multiple lines of computational evidence do not confidently suggest no impact.
BP5
No observation of this variant in a case with an alternate molecular basis for disease has been identified.
BP6
No reputable source has reported this variant as benign.
N/A · 7
PVS1 · PM3 · PM4 · PM5 · BP2 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
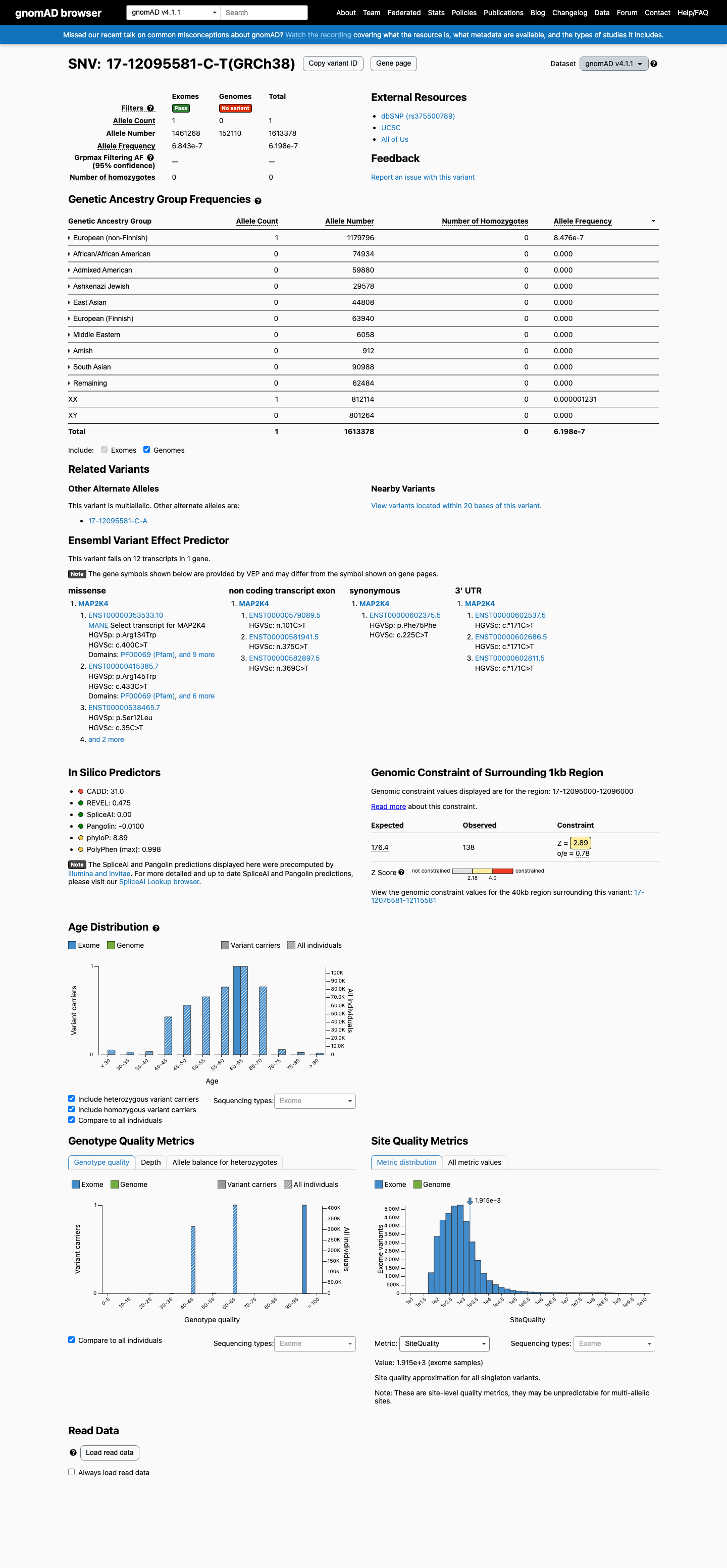
This variant is present in gnomAD v4.1 (AF= 6.19818e-07; MAF= 0.00006%, 1/1613378 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.47604e-07; MAF= 0.00008%, 1/1179796 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
6.2e-05%
· 1 / 1,613,378
0 hom
0 hom
European (non-Finnish) 1 / 1,179,796 |
8.5e-05% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.475. BayesDel score = 0.246892.
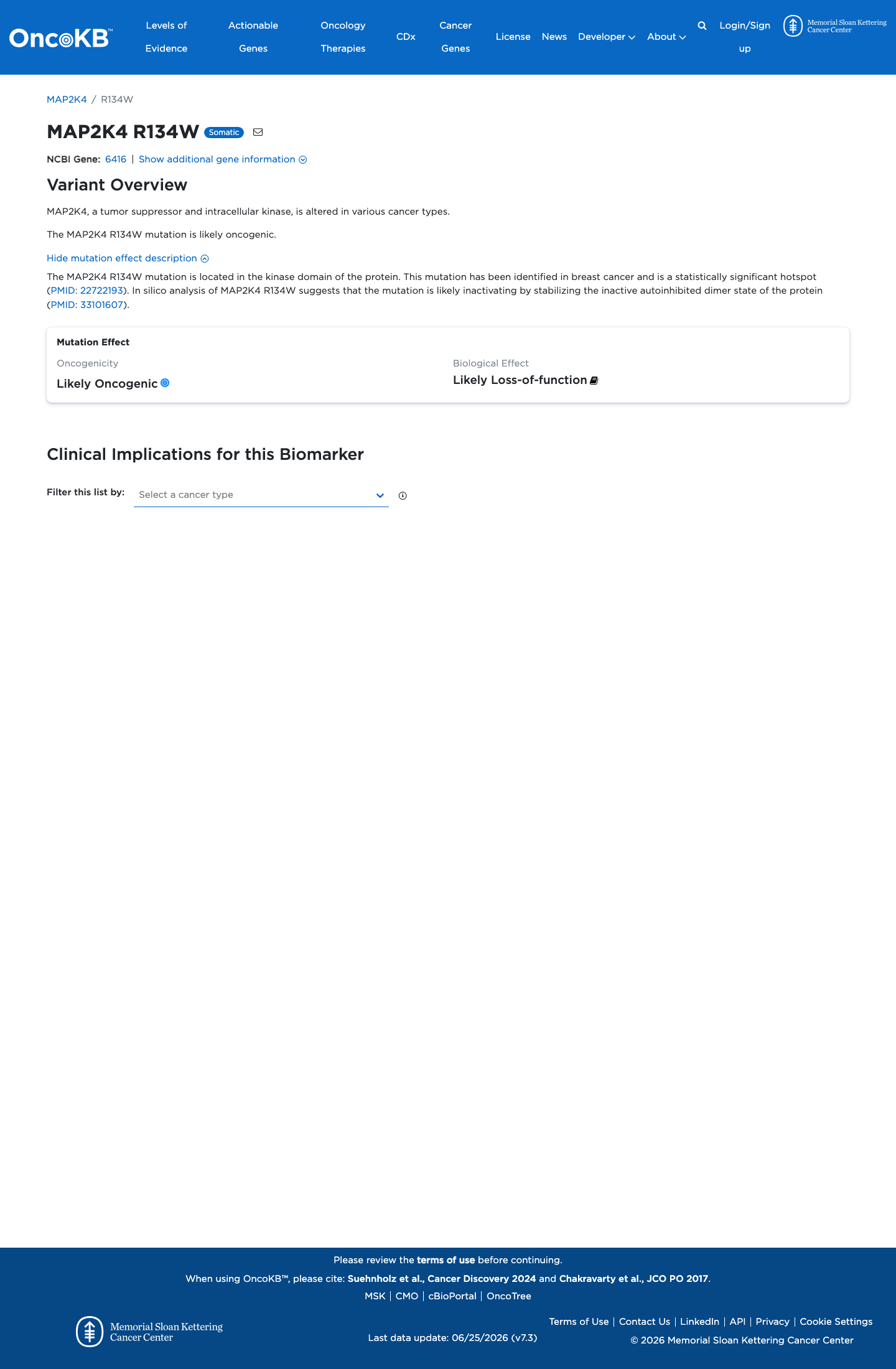
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.

COSMIC
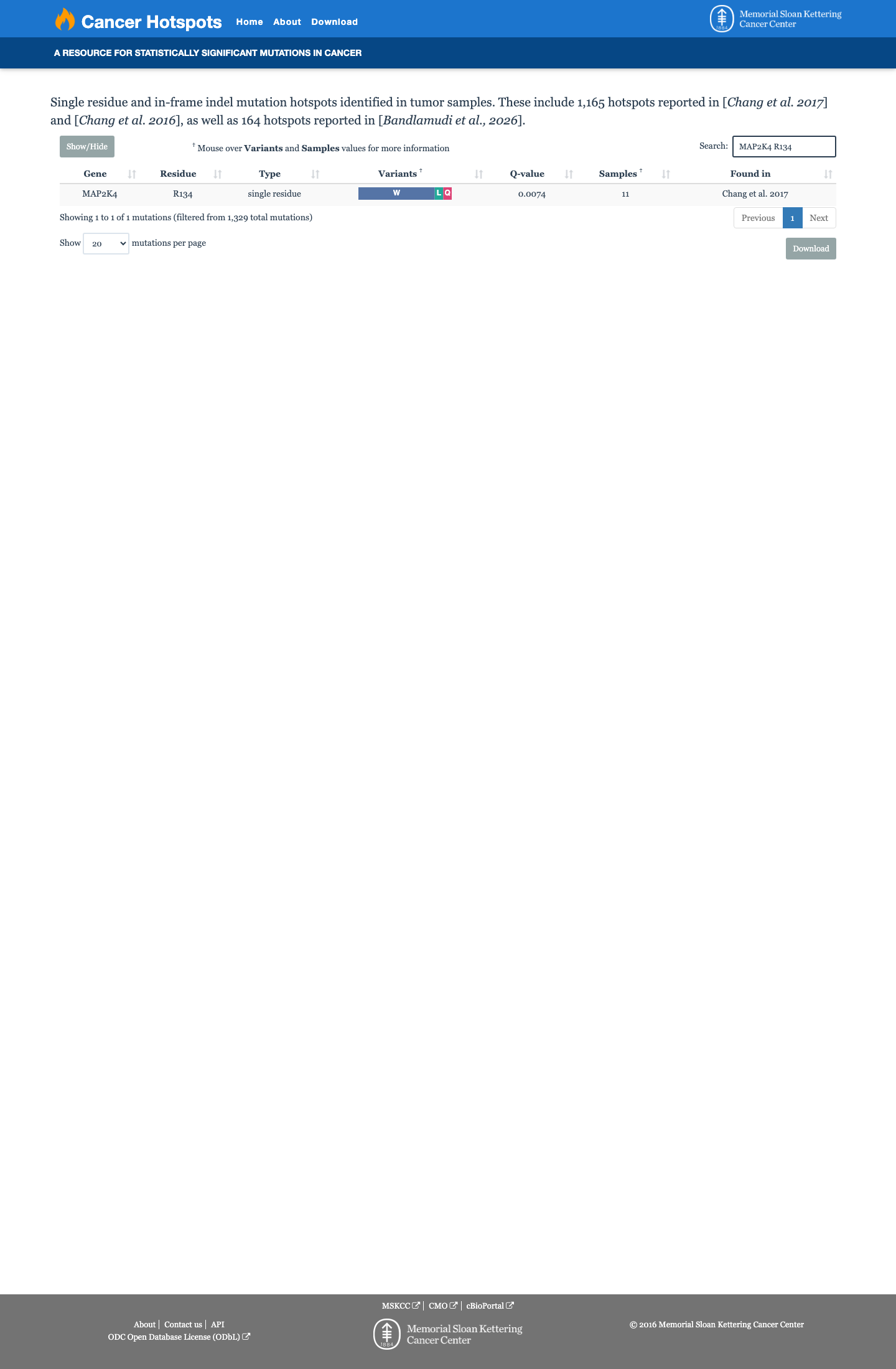
Cancer hotspots
Somatic evidence
Hotspot
COSMIC
This variant lies in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant lies in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 1 further PMID triaged but not cited — see Sources & References.
The autoinhibited state of MKK4: Phosphorylation, putative dimerization and R134W mutant studied by molecular dynamics simulations.
Searched
R134Wc.400C>TArg134TrpNM_003010.3
Found
Studied the conformational dynamics of autoinhibited MKK4 by microsecond-timescale all-atom MD simulations, including the R134W mutant. In the unphosphorylated homodimer, R134W introduces a π-π stacking interaction between TRP134 residues of opposing subunits that is absent in wild-type and distorts the N-lobe dimer interface. The mutation does not alter secondary structure but increases the angle variation between the αC-helix and activation segment helix, indicating altered dimer stability. The study is purely computational; no experimental functional assay (kinase activity, phosphorylation) was performed for R134W.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
Why
Used to support PM1 (hot-spot confirmation) and to document the absence of experimental functional data for PS3/BS3. Computational evidence alone does not meet PS3 or BS3 thresholds.
The most frequent MKK4 mutation R134W, where an arginine residue is replaced with a tryptophan, is located in the loop between β3-sheet and αC-helix. However, no functional data of the R134W mutation exist to date and its effect on MKK4's function is unclear.
Location Title; Introduction (para 2); Results section 3.2; Figures 5-7; Discussion · Context All-atom molecular dynamics simulations (40 μs total); OPLS3/OPLS3e force field; Desmond MD engine; in silico only · full text
Sources & reference links
Triaged references · 1 PMID not cited in assessment