NM_003073.4:c.1091_1093del (p.Lys364del) is an in-frame deletion in exon 8 of SMARCB1, removing a single lysine residue from the C-terminal domain alpha-helix.1 The variant has been identified in 9 independent individuals with Coffin-Siris syndrome, a rare autosomal dominant neurodevelopmental disorder characterized by intellectual disability, coarse facial features, and hypoplastic fifth digit nails.2 One confirmed de novo occurrence with both maternity and paternity confirmed has been reported in a CSS patient.3 The variant is essentially absent from large population databases (gnomAD v4.1: 1/1,614,188 alleles; gnomAD v2.1: absent), consistent with a rare pathogenic variant.4 Comprehensive functional studies by Valencia et al. (2019) directly demonstrated that the K364del variant disrupts SMARCB1 C-terminal domain binding to the nucleosome acidic patch, significantly attenuates mSWI/SNF complex nucleosome remodeling and ATPase activity, and impairs genome-wide DNA accessibility.5 In iPSC-derived neuronal differentiation models, heterozygous K364del resulted in significantly diminished neurite outgrowth and reduced neuron counts, with phenotypic rescue upon restoration of wild-type SMARCB1, establishing a direct causal link between this variant and neurodevelopmental deficits.6 Residue K364 is located within the SMARCB1 C-terminal alpha-helical domain (aa 357-377), a well-characterized functional domain that directly binds the nucleosome acidic patch and is essential for mSWI/SNF-mediated chromatin remodeling. This domain is a hotspot for CSS-associated mutations with no benign variation.7 The variant is classified as Pathogenic in ClinVar (Variation ID: 30201) by 8 clinical laboratories; however, the aggregate review status is 'criteria provided, single submitter' (1-star), which does not meet the 3-star threshold for PP5 application.8 Applying generic ACMG/AMP 2015 criteria: 2 strong (PS3, PS4), 4 moderate (PS2, PM1, PM2, PM4) pathogenic criteria are met. No benign criteria are met. This combination exceeds the threshold for Pathogenic classification (requires 2 strong OR 1 strong + ≥3 moderate).9
SMARCB1
Final classification
Unclassified
SMARCB1 c.1091_1093del · p.Lys364del
SMARCB1
NM_003073.4:c.1091_1093del (p.Lys364del) is an in-frame deletion in exon 8 of SMARCB1, removing a single lysine residue from the C-terminal domain alpha-helix.
Classification rationale
PS2PS3PS4PM1PM2PM4
Unclassified
SMARCB1 c.1091_1093del
· exon NC_000022.10
PS2 + PS3 + PS4 + PM1 + PM2 + PM4
→
Unclassified
1
PMID:22426308
2
PMID:22426308PMID:25081545PMID:31759698
3
PMID:22426308
5
PMID:31759698
6
PMID:31759698
7
PMID:31759698
9
generic_acmg_combination_rules
Gene diagram
· NM_003073.4 · variants mapped to exon structure
SMARCB1
NM_003073.4
Fetching transcript structure from UCSC…
Applied criteria · 6 applied · 9 assessed
Applied · 6
Strength
Supporting
Moderate
Strong
Very strong
✓
PS2
moderate
Pathogenic
A de novo occurrence of NM_003073.4:c.1091_1093del (p.Lys364del) with confirmed maternity and paternity was identified in one Coffin-Siris syndrome patient (subject 4) by Tsurusaki et al. 2012. Two additional CSS patients (subjects 21, 22) also harbor this variant but parental confirmation was not possible. One confirmed de novo in a disorder consistent with the variant's associated phenotype supports PS2 at moderate strength.
Subject 4: confirmed de novo p.Lys364del in CSS patient with both parental samples tested negativeSubjects 21 and 22: same variant but parental samples unavailable for de novo confirmationControl frequency 0/502 Japanese alleles
✓
PS3
strong
Pathogenic
Valencia et al. 2019 (PMID:31759698) directly tested the K364del variant in comprehensive functional assays. mSWI/SNF complexes containing K364del SMARCB1 exhibited significantly attenuated nucleosome remodeling activity and reduced ATPase activity on nucleosome substrates compared to wild-type. The K364del mutation completely disrupted SMARCB1 CTD binding to the nucleosome acidic patch. In SMARCB1-deficient MRT cell rescue experiments, K364del mutant complexes showed normal genome-wide targeting but substantially diminished DNA accessibility (ATAC-seq) and reduced nucleosome eviction (MNase-seq). Heterozygous K364del iPSCs demonstrated decreased DNA accessibility at pluripotency loci and impaired neuronal differentiation with significantly diminished neurite outgrowth, rescued by wild-type SMARCB1 restoration. The exact variant was directly tested with unequivocal deleterious functional effects across biochemical, genomic, and cellular differentiation assays.
Nucleosome remodeling (REAA): K364del mSWI/SNF complexes show significant attenuation of remodeling activityATPase activity: significantly reduced on nucleosome substrates but not on free DNACTD-nucleosome binding: K364del completely abrogates SMARCB1 CTD binding to mononucleosomes
✓
PS4
strong
Pathogenic
The p.Lys364del variant is the most recurrent SMARCB1 mutation in Coffin-Siris syndrome, identified in 9 independent CSS cases across published cohorts (Tsurusaki et al. 2012: 3 cases; Miyake et al. 2014: additional cases; Valencia et al. 2019: 9 total). The variant is essentially absent from population databases (gnomAD v4.1: 1/1,614,188 alleles, AF = 6.2e-07; gnomAD v2.1: absent). The extreme enrichment in affected individuals versus population controls meets PS4 at strong strength.
9 independent CSS cases with p.Lys364del across published studiesgnomAD v2.1: absentgnomAD v4.1: 1 allele in 1
✓
PM1
moderate
Pathogenic
The p.Lys364del variant is located in the SMARCB1 C-terminal domain (CTD; amino acids 357-377), a critical functional domain characterized by Valencia et al. 2019 as the nucleosome acidic patch binding domain. The CTD alpha-helix contains a dense cluster of basic residues required for mSWI/SNF complex-mediated nucleosome remodeling. This is a well-defined functional domain where missense and in-frame deletion mutations are enriched in both CSS and cancer, with no benign variation observed. PM1 is met at moderate strength based on the variant's location in this critical functional domain.
K364 is within the CTD alpha-helix (aa 357-377)the nucleosome acidic patch binding domainCTD is a well-characterized functional domain required for mSWI/SNF remodeling activity
✓
PM2
moderate
Pathogenic
The variant is essentially absent from large population databases. gnomAD v2.1: 0 alleles. gnomAD v4.1: 1 allele in 1,614,188 (AF = 6.2e-07, 0.000062%), well below the PM2 threshold of <0.1%. This extreme rarity in the general population supports PM2 at moderate strength.
gnomAD v2.1: absent (0 alleles)gnomAD v4.1: 1/1614
✓
PM4
moderate
Pathogenic
NM_003073.4:c.1091_1093del is an in-frame deletion of a single amino acid (p.Lys364del) in a non-repeat region of SMARCB1. The deletion removes a critical residue within the CTD nucleosome acidic patch binding domain, resulting in a protein length change with demonstrated functional consequences. PM4 is met at moderate strength.
In-frame single amino acid deletion (K364del) in exon 8Residue is in a non-repeatfunctionally critical region (CTD alpha-helix)
Assessed · not applied
Pathogenic
PP3
In silico pathogenicity prediction tools (REVEL, BayesDel) are not applicable for in-frame deletions and were not computed for this variant.
PP4
While patients with this variant present with classical Coffin-Siris syndrome features (severe intellectual disability, coarse facies, hypoplastic fifth digit nails), CSS is genetically heterogeneous — caused by mutations in at least six different BAF complex genes (SMARCB1, SMARCA4, SMARCA2, SMARCE1, ARID1A, ARID1B).
PP5
While ClinVar classifies this variant as Pathogenic (Variation ID: 30201) with submissions from 8 clinical laboratories, the aggregate review status is 'criteria provided, single submitter' (1-star).
Benign
BA1
The variant has an allele frequency of 6.2e-07 (0.000062%) in gnomAD v4.1, far below the BA1 threshold of >5%.
BS1
The variant has an allele frequency of 6.2e-07 (0.000062%) in gnomAD v4.1, far below the BS1 threshold of >0.3% for a dominant disorder.
BS2
A single allele is observed in gnomAD v4.1 (1/1,614,188).
BS3
Well-established functional studies (Valencia et al.
BP3
BP3 applies to in-frame deletions in repetitive regions without known function.
BP4
Computational predictors (REVEL, BayesDel) are not applicable for in-frame deletions.
N/A · 13
PVS1 · PS1 · PM3 · PM5 · PM6 · PP1 · PP2 · BS4 · BP1 · BP2 · BP5 · BP6 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
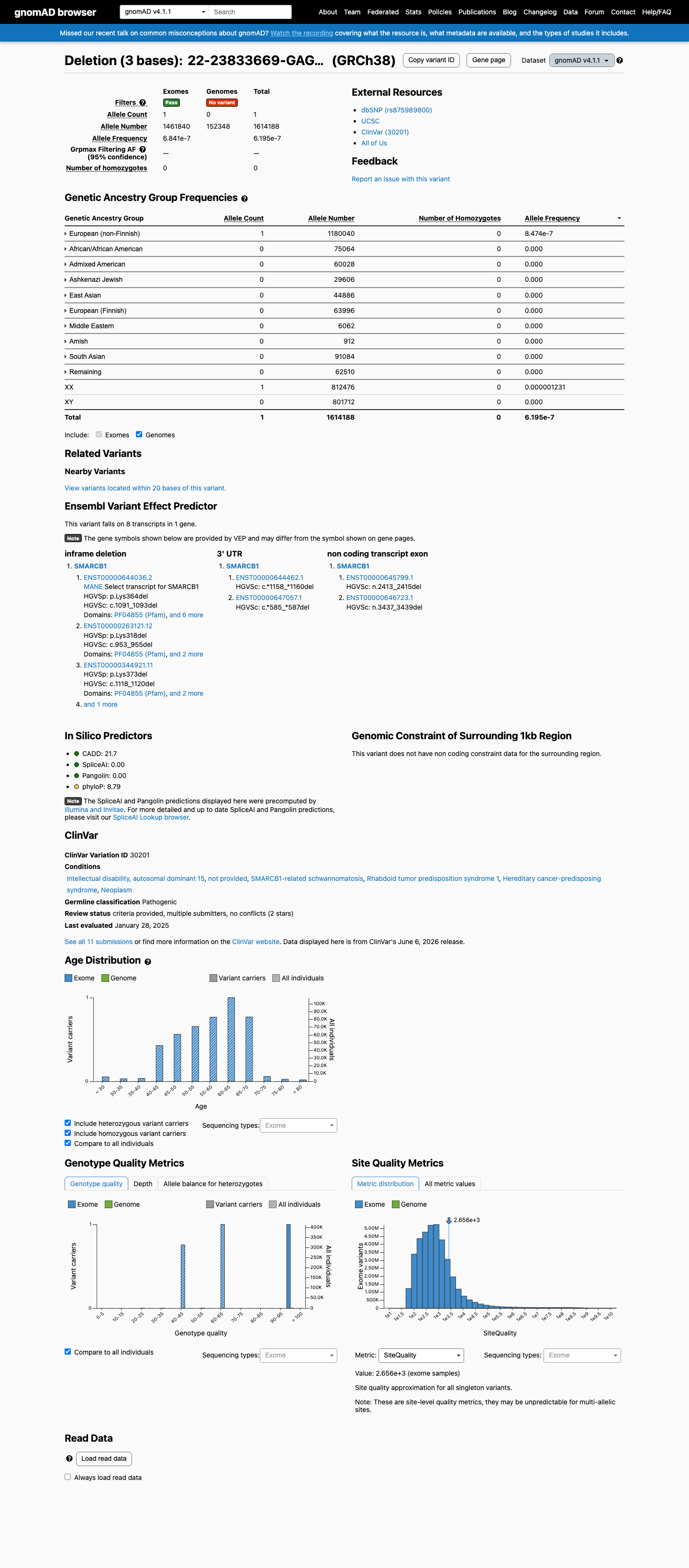
This variant is present in gnomAD v4.1 (AF= 6.19507e-07; MAF= 0.00006%, 1/1614188 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.47429e-07; MAF= 0.00008%, 1/1180040 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
6.2e-05%
· 1 / 1,614,188
0 hom
0 hom
European (non-Finnish) 1 / 1,180,040 |
8.5e-05% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.


COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
In progress — evidence not uploaded yet.
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
28481359 ↗
Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients.
ONCOKB
31759698 ↗
Recurrent SMARCB1 Mutations Reveal a Nucleosome Acidic Patch Interaction Site That Potentiates mSWI/SNF Complex Chromatin Remodeling.
ONCOKB
22426308 ↗
Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome.
CLINVAR
25168959 ↗
Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A.
CLINVAR
35101336 ↗
Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC).
CLINVAR
23637025 ↗
Clinical correlations of mutations affecting six components of the SWI/SNF complex: detailed description of 21 patients and a review of the literature.
CLINVAR
23619274 ↗
American College of Medical Genetics and Genomics technical standards and guidelines: microarray analysis for chromosome abnormalities in neoplastic disorders.
CLINVAR