Three independent de novo occurrences of c.1130G>A have been confirmed with parental testing in patients with Coffin-Siris syndrome / DOORS spectrum phenotype (Tsurusaki et al. 2012, Campeau et al. 2014).1 Functional studies (Valencia et al. 2019) demonstrate that the R377H variant completely disrupts SMARCB1 C-terminal domain binding to the nucleosome acidic patch, significantly attenuates mSWI/SNF chromatin remodeling activity, and reduces ATPase activity on nucleosome substrates.2 Codon 377 lies within the SMARCB1 C-terminal domain, a well-characterized functional domain essential for nucleosome binding and chromatin remodeling, and is a confirmed mutational hotspot in both germline and somatic disease contexts.3 The variant is absent from gnomAD v2.1 (0/216,632 alleles) and extremely rare in gnomAD v4.1 (2/1,593,918 alleles; AF=1.25e-6), meeting the population frequency criterion for PM2.4 Multiple in silico tools predict a deleterious effect (REVEL 0.868, BayesDel 0.543), supporting PP3.5 This variant is classified as Pathogenic in ClinVar by two clinical laboratories, supporting PP5.6 This variant has been observed in at least 12 affected individuals across multiple publications in both germline (CSS, DOORS) and somatic (meningioma, ameloblastoma, gastric cancer) disease contexts.7
SMARCB1
Final classification
Pathogenic
SMARCB1 c.1130G>A · p.Arg377His
SMARCB1
Three independent de novo occurrences of c.1130G>A have been confirmed with parental testing in patients with Coffin-Siris syndrome / DOORS spectrum phenotype (Tsurusaki et al. 2012, Campeau et al. 2014).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PS2 strong, PS3 moderate, PS4 supporting, PM1 moderate, PM2 supporting, PP3 supporting, PP5 supporting; combination = 1 strong + 2 moderate + 4 supporting, which maps to Pathogenic.
Classification rationale
PS2PS3PS4PM1PM2PP3PP5
Pathogenic
SMARCB1 c.1130G>A
PS2 + PS3 + PS4 + PM1 + PM2 + PP3 + PP5
→
Pathogenic
Gene diagram
· NM_003073.4 · variants mapped to exon structure
SMARCB1
NM_003073.4
Fetching transcript structure from UCSC…
Applied criteria · 7 applied · 12 assessed
Applied · 7
Strength
Supporting
Moderate
Strong
Very strong
✓
PS2
strong
Pathogenic
Three independent de novo occurrences of c.1130G>A (p.Arg377His) have been confirmed with parental testing across two publications. Tsurusaki et al. (2012, PMID:22426308) identified a de novo c.1130G>A mutation in a CSS patient (subject 11), confirmed by Sanger sequencing and absent from 500 control chromosomes. Campeau et al. (2014, PMID:25169651) identified two additional patients with the same de novo SMARCB1 c.1130G>A mutation, both confirmed by Sanger sequencing. All three patients presented with phenotypes consistent with Coffin-Siris syndrome / DOORS syndrome spectrum. Multiple independent de novo observations with confirmed parentage in affected individuals with consistent phenotype support PS2 at strong level.
Subject 11 (PMID:22426308): c.1130G>Ap.Arg377Hisconfirmed de novo
✓
PS3
moderate
Pathogenic
Valencia et al. (2019, PMID:31759698) directly tested the R377H variant in a comprehensive biochemical and functional characterization of the SMARCB1 C-terminal domain. The R377H mutation completely disrupted nucleosome binding by the SMARCB1 CTD peptide in gel-shift assays. Both K364del and R377H mutations led to a reduction in nucleosome crosslinking across experiments. Complexes containing SMARCB1 CTD mutant variants (including R377H) exhibited significant attenuation in nucleosome remodeling activity and a significant reduction in ATPase activity when bound to nucleosome substrates. ATAC-seq in rescue experiments showed decreased genome-wide chromatin accessibility with mutant SMARCB1 relative to wild-type. This is variant-specific functional evidence from a single systematic study; moderate strength is assigned per PS3 calibration (single study with systematic characterization directly testing the variant).
R377H completely disrupted nucleosome binding by SMARCB1 CTD in gel-shift assays (Figure 2B)R377H mutation led to reduction in nucleosome crosslinking across experiments (Figure 3B-D)mSWI/SNF complexes with R377H showed significant attenuation in nucleosome remodeling activity (Figure 1F-G)
✓
PS4
supporting
Pathogenic
This variant has been observed in multiple affected individuals across several publications in both germline and somatic contexts. Tsurusaki et al. (2012, PMID:22426308) reported 1 de novo germline case with CSS; Kosho et al. (2014, PMID:25168959) reviewed an additional germline case (Y11) with CSS; Campeau et al. (2014, PMID:25169651) reported 2 additional de novo germline cases; Schmitz et al. (2001, PMID:11161377) identified 4 somatic cases in meningiomas; Brown et al. (2014, PMID:24993163) identified 3 somatic cases in ameloblastomas; Kim et al. (2013, PMID:23196062) identified 1 somatic case in gastric cancer. The variant is absent from gnomAD v2.1 (0/216,632) and present at only 2/1,593,918 alleles (AF=1.25e-6, grpmax FAF=2.8e-7) in gnomAD v4.1. The aggregate case count supports enrichment in affected individuals relative to the general population, meeting PS4 at supporting level.
1 de novo germline CSS case (PMID:22426308)1 germline CSS case Y11 (PMID:25168959)2 de novo germline DOORS/CSS cases (PMID:25169651)
✓
PM1
moderate
Pathogenic
Codon 377 lies within the SMARCB1 C-terminal domain (CTD, aa 351-385), a well-characterized functional domain that directly binds the nucleosome acidic patch and is essential for mSWI/SNF complex-mediated chromatin remodeling activity (PMID:31759698). Schmitz et al. (2001, PMID:11161377) identified this codon as a recurrent mutational hotspot in meningiomas. The variant falls within the highly conserved coiled coil domain at the C-terminus. Both germline (CSS, DOORS) and somatic (meningioma, ameloblastoma, gastric cancer) mutations cluster at this residue. Cancerhotspots.org identifies this as a statistically significant hotspot residue. PM1 at moderate is supported by a well-characterized functional domain with a confirmed mutational hotspot.
Residue 377 located in SMARCB1 C-terminal domain (CTD) coiled coilcritical for nucleosome acidic patch bindingPMID:11161377 identifies codon 377 as a mutational hotspot in meningiomas
✓
PM2
supporting
Pathogenic
This variant is absent from gnomAD v2.1 exomes (0/216,632 alleles) and present at extremely low frequency in gnomAD v4.1 (2/1,593,918 alleles, AF=1.25e-6, grpmax FAF=2.8e-7), well below the 0.1% threshold for PM2. The near-complete absence from large population databases supports PM2. Supporting strength is assigned because 2 alleles were detected in v4.1, indicating the variant is not universally absent, though the grpmax filtering allele frequency of 2.8e-7 confirms extreme rarity.
gnomAD v2.1: 0/216632 alleles (AF=0)gnomAD v4.1: 2/1
✓
PP3
supporting
Pathogenic
Multiple in silico prediction tools support a deleterious effect for this missense variant. REVEL score of 0.868 is strongly predictive of pathogenicity. BayesDel score of 0.543 is above the deleterious threshold. SpliceAI predicts no splicing impact (max delta 0.00), so the computational evidence is specific to the amino acid substitution rather than a cryptic splice effect. PP3 at supporting level is appropriate given converging in silico evidence.
REVEL score: 0.868 (deleterious)BayesDel score: 0.543 (deleterious)SpliceAI max delta: 0.00 (no cryptic splice effect)
✓
PP5
supporting
Pathogenic
This variant is classified as Pathogenic in ClinVar (Variation ID: 30203) by two clinical testing laboratories (Labcorp Genetics/Invitae and Ambry Genetics), both with criteria provided. Although the review status is 1★ (single submitter level), the classification is consistent across submitters and is supported by published literature. Under generic ACMG rules, a reputable clinical laboratory classification of Pathogenic supports PP5 at supporting level.
ClinVar Variation ID 30203: PathogenicTwo clinical laboratories: Labcorp Genetics (SCV001411715) and Ambry Genetics (SCV001178478)Both submitters provided criteria
Assessed · not applied
Pathogenic
PP1
No segregation data are available for this variant.
PP2
PP2 requires a computational gene-level missense constraint metric (Z-score) that is not available in the case data.
PP4
PP4 requires that the patient's phenotype is highly specific for a disease with a single genetic etiology.
Benign
BA1
The maximum allele frequency of this variant (gnomAD v4.1: AF=1.25e-6, grpmax FAF=2.8e-7) is far below the BA1 threshold of >1%.
BS1
The allele frequency of this variant (gnomAD v4.1: AF=1.25e-6) is far below the BS1 threshold of >0.3%.
BS2
No evidence was found of this variant being observed in healthy adults.
BS3
Functional studies (PMID:31759698) demonstrate a deleterious effect of the R377H variant on SMARCB1 CTD nucleosome binding, crosslinking, chromatin remodeling activity, and ATPase activity.
BS4
No evidence of lack of segregation with disease was identified.
BP2
No evidence was found of this variant being observed in trans with a known pathogenic SMARCB1 variant.
BP4
Multiple in silico tools (REVEL: 0.868; BayesDel: 0.543) predict a deleterious effect.
BP5
This variant has been observed in multiple affected individuals with a consistent disease phenotype across independent publications (CSS, DOORS).
BP6
ClinVar classifies this variant as Pathogenic (Variation ID: 30203) by two clinical laboratories.
N/A · 9
PVS1 · PS1 · PM3 · PM4 · PM5 · PM6 · BP1 · BP3 · BP7
Research & evidence
Population frequency · supports pathogenic
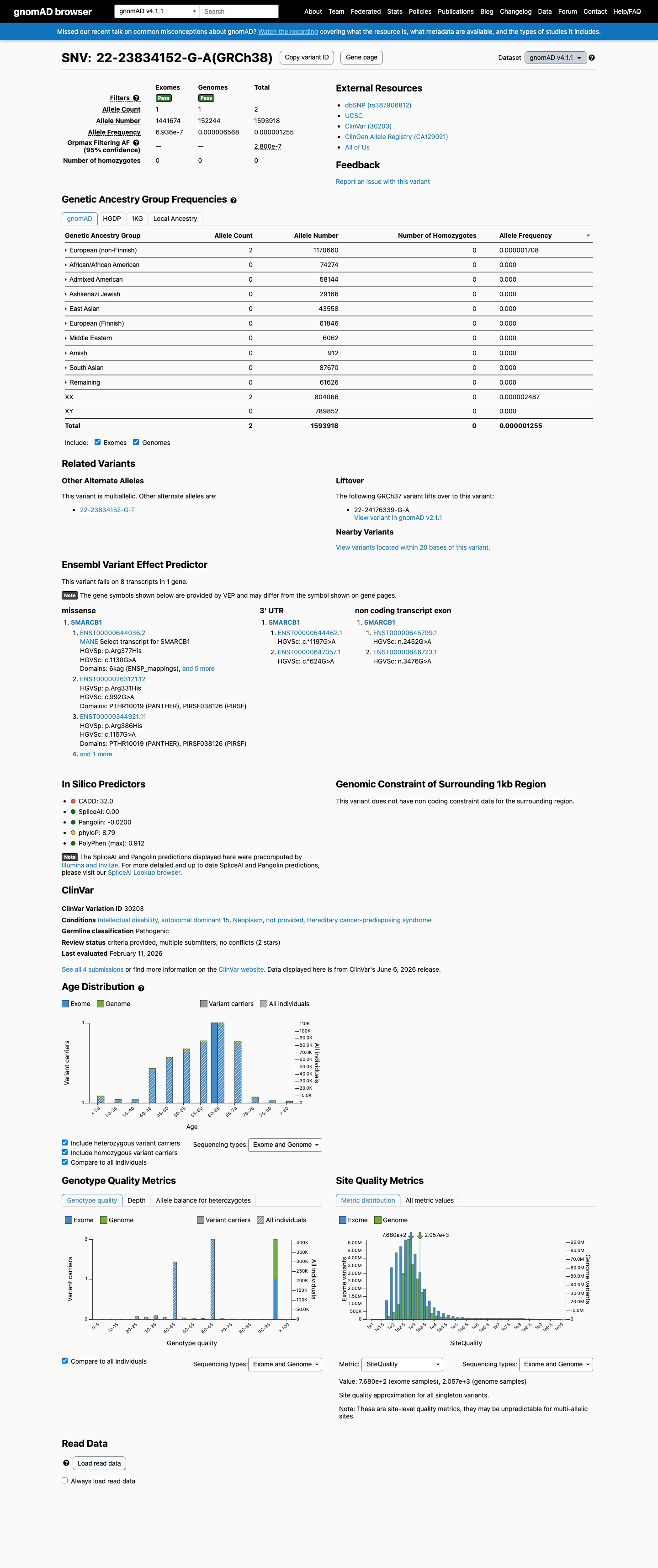
gnomAD v4.1
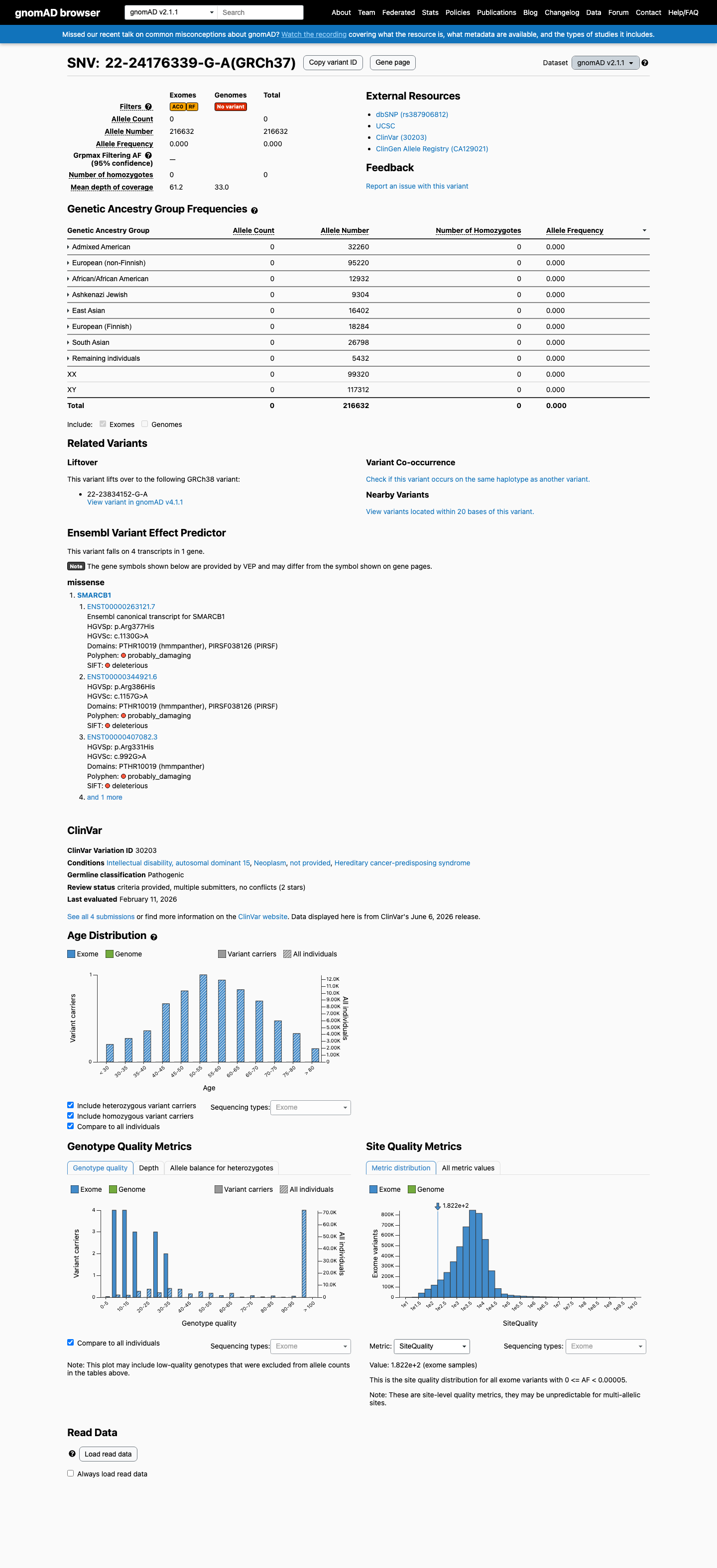
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 1.25477e-06; MAF= 0.00013%, 2/1593918 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 1.70844e-06; MAF= 0.00017%, 2/1170660 alleles, homozygotes = 0); grpmax FAF= 2.8e-07.
v2.1
This variant is present in gnomAD v2.1 (AF= 0; MAF= 0.00000%, 0/216632 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0; MAF= 0.00000%, 0/12932 alleles, homozygotes = 0).
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00013%
· 2 / 1,593,918
0 hom · FAF 2.8e-05%
0 hom · FAF 2.8e-05%
European (non-Finnish) 2 / 1,170,660 |
0.00017% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / 216,632
0 hom
0 hom
Not observed in any ancestry group.
+ 8 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
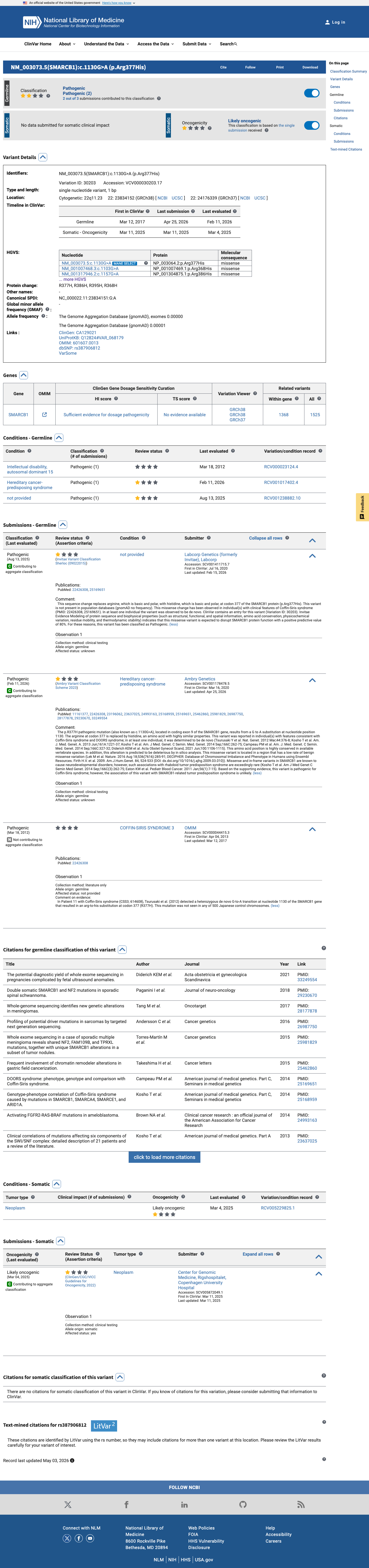
ClinVar
This variant has been reported in ClinVar as Pathogenic (2 clinical laboratories). (ClinVarID = 30203)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.868. BayesDel score = 0.542711.
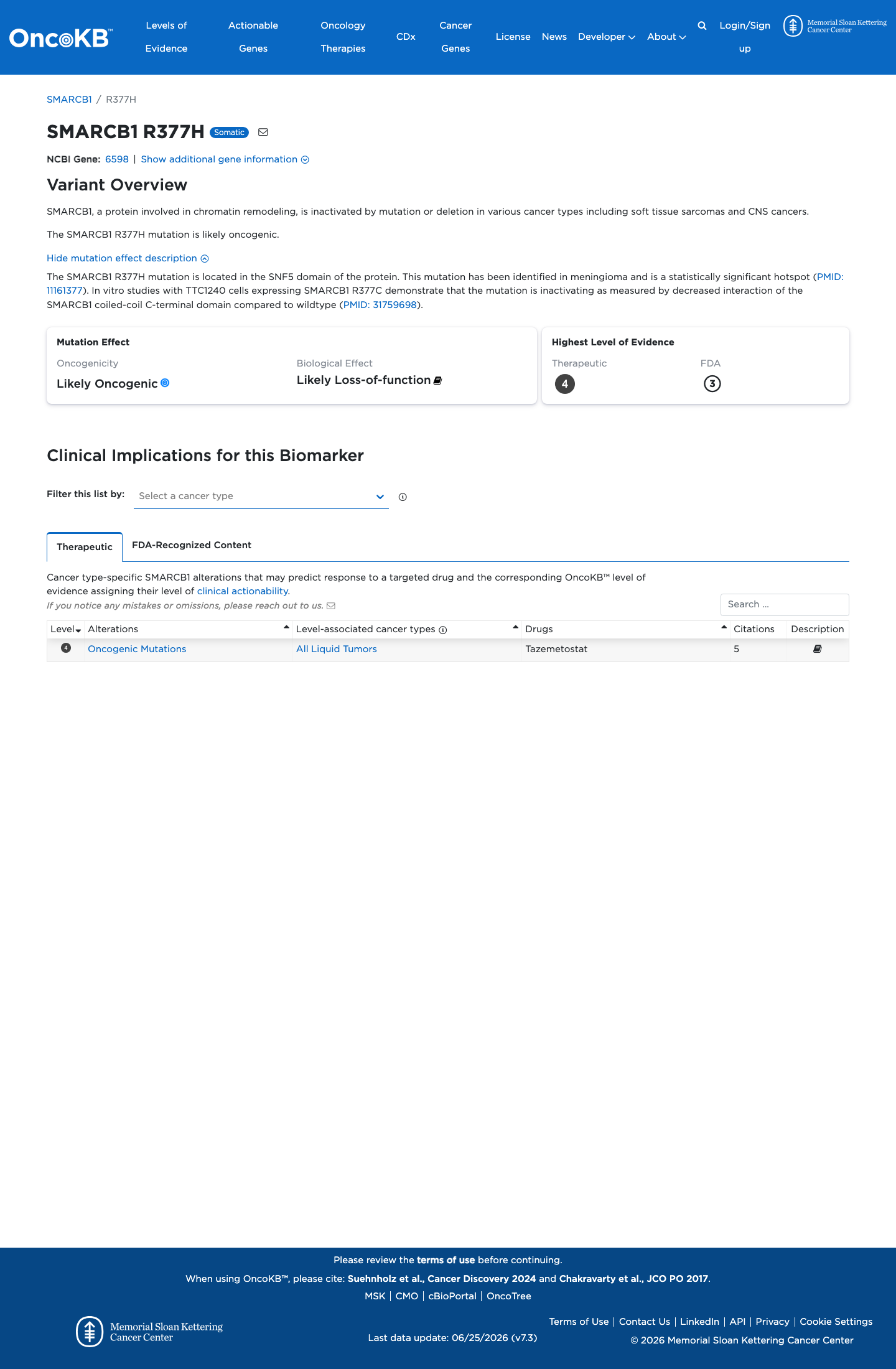
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.

COSMIC
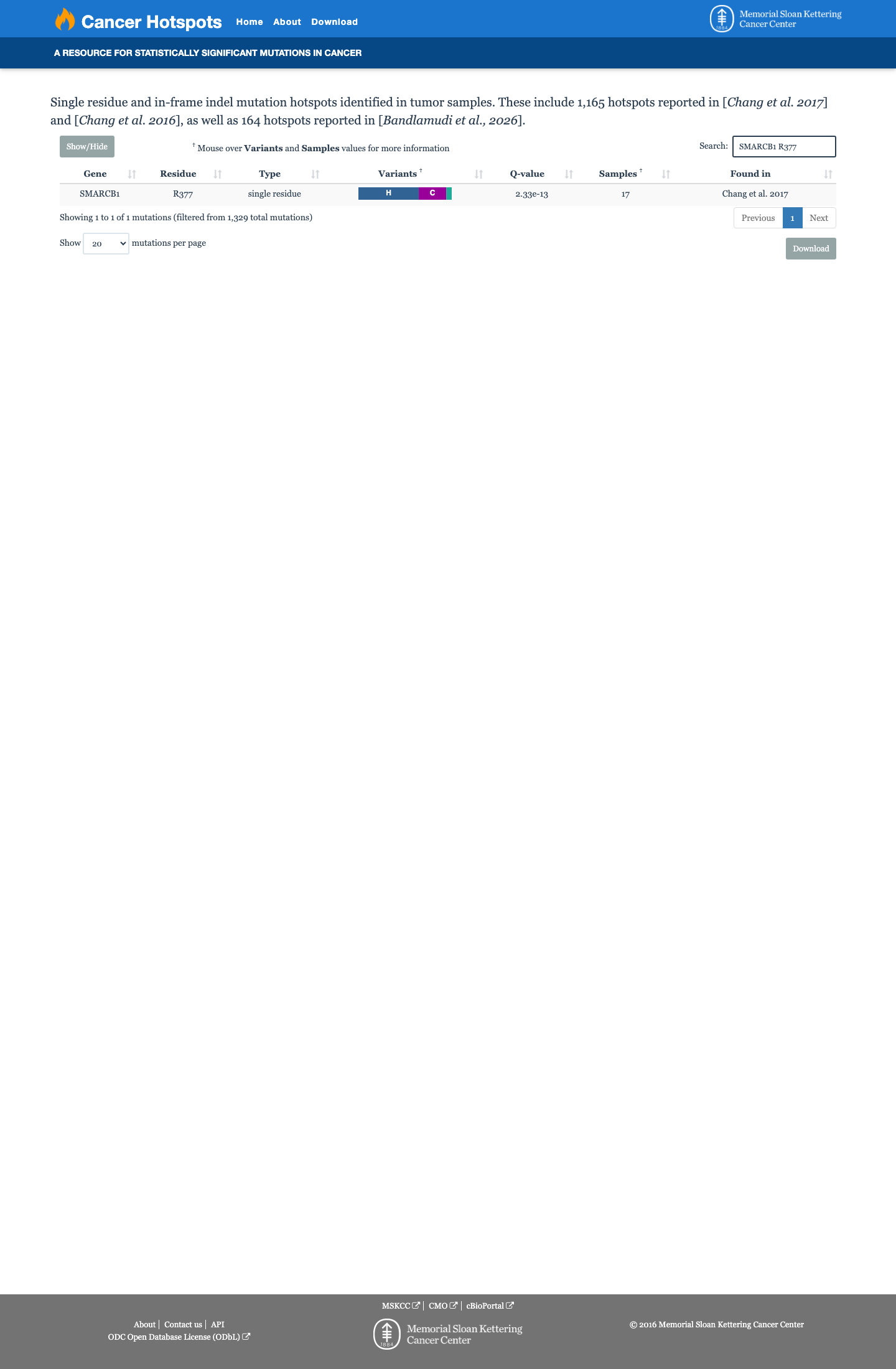
Cancer hotspots
Somatic evidence
Hotspot
COSMIC
This variant lies in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant lies in a statistically significant hotspot.
Literature · how each cited paper was used
7papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 1 further PMID triaged but not cited — see Sources & References.
INI1 mutations in meningiomas at a potential hotspot in exon 9.
Searched
c.1130G>Ap.Arg377HisR377H1130Arg to Hiscodon 377
Found
Identified the somatic c.1130G>A (p.Arg377His) mutation in 4 of 126 meningiomas. The alteration was not observed in 104 healthy individuals or 200 other brain tumors, confirming somatic origin. The Arg377His change falls within the highly conserved C-terminal coiled coil domain and was characterized as a mutational hotspot in exon 9. No functional data on the effect of the mutation were available at the time of publication.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
→PS4 supports · met
Why
Confirms codon 377 as a mutational hotspot and provides independent somatic case observations; referenced in PM1 and PS4 assessments.
The alteration was characterized by an G to A transition in the nucleotide 1130 of the coding sequence resulting in an missense mutation of Arg to His in codon 377.
Location Results and Discussion; Figure 1 (lower panel) · Context SSCP analysis and Sanger sequencing of meningioma tumor DNA vs matched blood DNA · full text
Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome.
Searched
c.1130G>Ap.Arg377HisArg3771130
Found
Identified c.1130G>A (p.Arg377His) as a de novo heterozygous germline mutation in subject 11 with typical Coffin-Siris syndrome, confirmed by Sanger sequencing and absent from 500 Japanese control chromosomes. This was the first report of SMARCB1 mutations causing CSS, establishing the gene-disease association. The mutation was non-truncating, suggesting gain-of-function or dominant-negative effects rather than haploinsufficiency.
Variant
✓ Names this variant — characterised directly
Applied to
→PS2 supports · met
→PS4 supports · met
Why
Confirmed de novo germline variant with CSS phenotype; referenced in PS2 (strong) and PS4 assessments.
two de novo heterozygous mutations of SMARCB1 were found in 2 affected individuals (c.1130G>A (p.Arg377His) and c.1091_1093del AGA (p.Lys364del))
Location Results paragraph 1; Table 1 (Subject 11) · Context Whole-exome sequencing followed by Sanger confirmation; parental testing confirmed de novo · full text
Comprehensive DNA methylation and extensive mutation analyses reveal an association between the CpG island methylator phenotype and oncogenic mutations in gastric cancers.
Searched
c.1130G>Ap.Arg377HisR377H1130
Found
Identified the somatic c.1130G>A (p.Arg377His) mutation in SMARCB1 in a gastric cancer sample (S5TP) alongside a KRAS G13D mutation. This was the first report of SMARCB1 mutations in gastric cancer. The mutation was confirmed somatic by dideoxy sequencing of matched non-cancerous tissue.
Variant
✓ Names this variant — characterised directly
Applied to
→PS4 supports · met
Why
Provides an additional somatic case observation; referenced in PS4 assessment.
SMARCB1 50 56 c.1130G>A p.Arg377His
Location Table 1 (Sample S5TP); Results section 3.2; Discussion section · Context Ion AmpliSeq Cancer Hotspot Panel (55 genes) on 30 gastric cancers; confirmed with dideoxy sequencing · full text
Activating FGFR2-RAS-BRAF mutations in ameloblastoma.
Searched
R377HArg377SMARCB1377
Found
Identified SMARCB1 R377H as a somatic mutation in 3 of 50 ameloblastomas using the Ion AmpliSeq Cancer Hotspot Panel, all confirmed by Sanger sequencing of matched normal tissue. SMARCB1 (INI-1) immunohistochemistry showed retained protein expression, indicating no loss of protein stability. SMARCB1 mutations co-occurred with activating FGFR2-RAS-BRAF mutations and were suggested to function as secondary mutations in ameloblastoma pathogenesis.
Variant
✓ Names this variant — characterised directly
Applied to
→PS4 supports · met
Why
Provides three additional somatic case observations; INI-1 IHC retained, indicating preserved protein expression but not contradicting the dominant-negative mechanism; referenced in PS4 assessment.
SMARCB1 R377H (3). All of these missense mutations were previously described in other neoplasms.
Location Results section 'Other mutations'; Supplementary Figure S1 and Table S1; Supplementary Figure S2B · Context Ion AmpliSeq Cancer Hotspot Panel v2 on 50 ameloblastomas with Sanger confirmation and IHC for INI-1 · full text
Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A.
Searched
c.1130G>Ap.Arg377HisArg3771130
Found
Reviewed genotype-phenotype correlation of CSS caused by SMARCB1 mutations. Patient Y11, a 7-year-old female with the c.1130G>A (p.Arg377His) mutation, exhibited severe neurodevelopmental deficits including severe intellectual disability, seizures, CNS structural abnormalities, scoliosis, sparse scalp hair, thick eyebrows, wide nasal bridge, cleft palate, and hypoplastic 5th fingernails/toenails. The mutation clustered within exons 8-9 at the C-terminus around the SNF5 domain.
Variant
✓ Names this variant — characterised directly
Applied to
→PS4 supports · met
Why
Provides detailed phenotype for an additional germline case; referenced in PS4 assessment.
the p.Arg377His was found in Y11 and also found as a somatic mutation in four meningiomas [Schmitz et al., 2001]
Location Table I (Patient Y11); Discussion section · Context Clinical review with genotype-phenotype correlation across 13 SMARCB1-mutated CSS patients · full text
DOORS syndrome: phenotype, genotype and comparison with Coffin-Siris syndrome.
Searched
c.1130G>Ap.Arg377HisR377H1130
Found
Identified the same de novo SMARCB1 c.1130G>A (p.Arg377His) mutation in two patients clinically diagnosed with DOORS syndrome. Both patients had Dandy-Walker malformation, retinal anomalies, respiratory difficulties, scoliosis, rocker bottom feet, and absence of seizures — distinguishing features from TBC1D24-associated DOORS syndrome. One patient died in the first year of life. Authors noted phenotypic overlap with Coffin-Siris syndrome but left the precise clinical classification undecided.
Variant
✓ Names this variant — characterised directly
Applied to
→PS2 supports · met
→PS4 supports · met
Why
Two additional de novo occurrences with consistent neurodevelopmental phenotype; referenced in PS2 (strong) and PS4 assessments.
we identified two patients with a de novo SMARCB1 c.1130G>A, p.Arg377His mutation, confirmed by Sanger sequencing
Location Abstract; Results (Genotypes section); Table I · Context Whole exome sequencing with Sanger confirmation; parental testing confirmed de novo · full text
Recurrent SMARCB1 Mutations Reveal a Nucleosome Acidic Patch Interaction Site That Potentiates mSWI/SNF Complex Chromatin Remodeling.
Searched
R377HArg377377c.1130
Found
Directly tested the R377H variant alongside other CSS-associated SMARCB1 CTD mutations. R377H completely disrupted SMARCB1 CTD binding to mononucleosomes in gel-shift assays. Both K364del and R377H mutations led to a reduction in nucleosome crosslinking. mSWI/SNF complexes containing SMARCB1 CTD variants exhibited significant attenuation in nucleosome remodeling activity and reduced ATPase activity on nucleosome substrates. ATAC-seq in SMARCB1-deficient MRT cells showed decreased genome-wide chromatin accessibility with mutant rescue compared to wild-type.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
→PS3 supports · met
Why
Variant-specific functional data confirmed deleterious effect on nucleosome binding and chromatin remodeling; referenced in PS3 (moderate) and PM1 assessments.
both K364del and R377H mutations led to a reduction in crosslinking across experiments (Figure 3B-D)
Location Results; Figures 1F-G, 1H-J, 2B, 3B-D; Figure 4F-G · Context NMR spectroscopy, photocrosslinking with diazirine probes, nucleosome remodeling restriction enzyme accessibility assays (REAA), ATPase activity assays in HEK-293T SMARCB1-KO cells, ATAC-seq in TTC1240 and G401 MRT cell lines · full text
Sources & reference links
Triaged references · 1 PMID not cited in assessment
23619274 ↗
American College of Medical Genetics and Genomics technical standards and guidelines: microarray analysis for chromosome abnormalities in neoplastic disorders.
CLINVAR