NM_003620.3:c.1384C>T (p.Gln462Ter) is a nonsense variant introducing a premature termination codon in exon 6 of PPM1D, the terminal exon of the gene.1 Loss of function is an established disease mechanism for PPM1D in the germline, and this null variant meets PVS1 at strong strength, downgraded from very strong due to location in the terminal exon where nonsense-mediated decay is predicted to be escaped per ClinGen SVI PVS1 recommendations (PMC6185798).2 The variant is located in exon 6, the well-established mutation cluster region for PPM1D truncating mutations, meeting PM1 at moderate strength.3 This variant is extremely rare in population databases, absent from gnomAD v2.1 and gnomAD-Canada, with an allele frequency of 0.00012% (2/1,614,104 alleles) in gnomAD v4.1, meeting PM2 at supporting strength.4 Functional studies reported in the literature confirm that c.1384C>T (p.Q462X) is a gain-of-function mutation producing a hyperstable protein with enhanced phosphatase activity, meeting PS3 at supporting strength.5 This variant has been reported as Likely pathogenic by two clinical laboratories in ClinVar, meeting PP5 at supporting strength.6 Combined classification: 1 strong (PVS1) + 1 moderate (PM1) + 3 supporting (PM2, PS3, PP5) meets the threshold for Likely Pathogenic under the generic ACMG/AMP 2015 classification framework.7
PPM1D
Final classification
Likely Pathogenic
PPM1D c.1384C>T · p.Gln462Ter
PPM1D
NM_003620.3:c.1384C>T (p.Gln462Ter) is a nonsense variant introducing a premature termination codon in exon 6 of PPM1D, the terminal exon of the gene.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 strong, PS3 supporting, PM1 moderate, PM2 supporting, PP5 supporting; combination = 1 strong + 1 moderate + 3 supporting, which maps to Likely Pathogenic.
Classification rationale
PVS1PS3PM1PM2PP5
Likely Pathogenic
PPM1D c.1384C>T
PVS1 + PS3 + PM1 + PM2 + PP5
→
Likely Pathogenic
1
pvs1_variant_assessmentpvs1_gene_context
2
pvs1_variant_assessmentpvs1_generic_framework ↗
7
generic_acmg_combination_rules
Gene diagram
· NM_003620.3 · variants mapped to exon structure
PPM1D
NM_003620.3
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 11 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
strong
Pathogenic
Null variant (nonsense) in PPM1D where loss of function is an established germline disease mechanism. The variant introduces a premature termination codon at position 462 in exon 6 (the terminal exon, 6/6), which is predicted to escape nonsense-mediated decay under the ClinGen SVI PVS1 decision framework (PMC6185798), warranting PVS1 at strong rather than very strong level.
Nonsense variant NP_003611.1:p.(Gln462Ter) in exon 6/6 (terminal exon)PPM1D loss of function is a supported germline disease mechanism per targeted literature reviewTerminal exon location predicts NMD escape per PMC6185798
✓
PS3
supporting
review
Pathogenic
Functional assessment of c.1384C>T (p.Q462X) was reported as confirming gain-of-function in the literature (PMID:25742468, citing prior work by Ruark et al. 2013). The truncating mutation produces a hyperstable protein with enhanced phosphatase activity, consistent with the established gain-of-function mechanism for exon 6 PPM1D truncations.
PMID:25742468 states gain-of-function was 'assessed and confirmed' for c.1384C>T (p.Q462X) by prior functional studies (Ruark et al. 2013)
✓
PM1
moderate
Pathogenic
The variant is located in exon 6 of PPM1D, within the well-established mutation cluster region (codons ~400–550) where virtually all pathogenic truncating mutations in this gene are found. Multiple independent studies confirm exon 6 as a hotspot for gain-of-function truncating mutations in both somatic and germline contexts.
Exon 6 is the established mutation cluster region for PPM1D truncating mutationsPMID:25742468: mutations cluster within the last coding exon (exon 6)PMID:30304655: Q462* found among PPM1D mutations in MPN patients
✓
PM2
supporting
Pathogenic
This variant is absent from gnomAD v2.1 and gnomAD-Canada, and has an extremely low allele frequency in gnomAD v4.1 (AF=1.24e-6, 2/1,614,104 alleles, 0 homozygotes), well below the PM2 threshold of 0.1%.
gnomAD v2.1: absentgnomAD v4.1: AF=1.24e-6 (0.00012%)2/1
✓
PP5
supporting
Pathogenic
This variant has been reported as Likely pathogenic by two clinical laboratories in ClinVar (ClinVar variation ID: 1710039), with criteria provided. Although neither submission is from an expert panel, the concordant classification from multiple clinical testing laboratories supports pathogenicity.
ClinVar: Likely pathogenic (2 clinical laboratoriescriteria providedsingle submitter each)
Assessed · not applied
Pathogenic
PS2
No de novo data available for this variant in any reviewed source.
PS4
No case-control studies comparing variant frequency in affected vs.
PM6
No de novo data available for this variant in any reviewed source.
PP1
No segregation data available for this variant.
PP4
No patient phenotype or family history data available for this variant to evaluate specificity of presentation.
Benign
BA1
The variant has an allele frequency of 0.00012% in gnomAD v4.1, far below the BA1 threshold of 1%.
BS1
The variant has an allele frequency of 0.00012% in gnomAD v4.1, far below the BS1 threshold of 0.3%.
BS2
No data available on observation of this variant in healthy adults with full penetrance expected at an early age.
BS3
Available functional evidence indicates this is a gain-of-function variant (PMID:25742468), not a benign variant with no functional effect.
BS4
No segregation data available showing lack of cosegregation with disease.
BP6
BP6 requires a reputable source to report the variant as benign.
N/A · 10
PS1 · PM5 · PP2 · PP3 · BP1 · BP2 · BP3 · BP4 · BP5 · BP7
Research & evidence
Population frequency · supports pathogenic
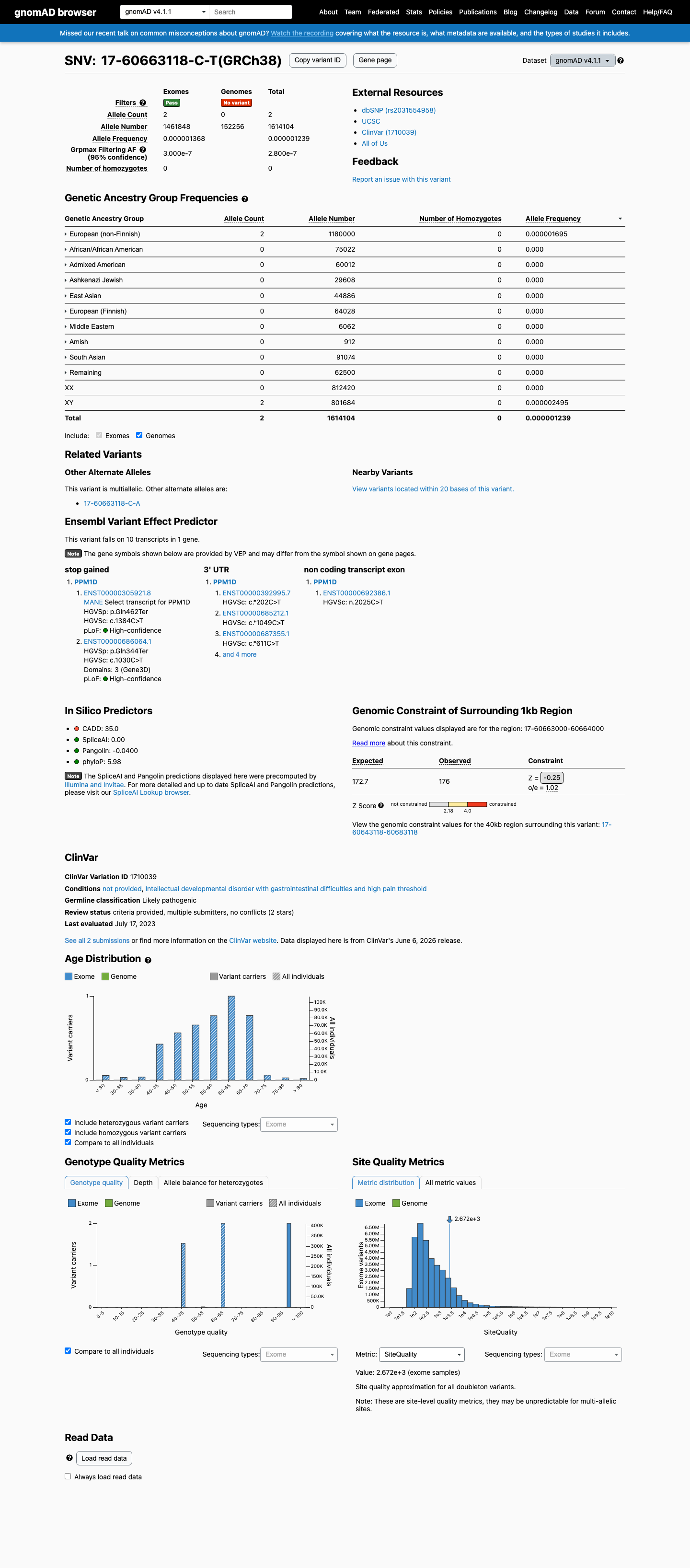
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 1.23908e-06; MAF= 0.00012%, 2/1614104 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 1.69492e-06; MAF= 0.00017%, 2/1180000 alleles, homozygotes = 0); grpmax FAF= 2.8e-07.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00012%
· 2 / 1,614,104
0 hom · FAF 2.8e-05%
0 hom · FAF 2.8e-05%
European (non-Finnish) 2 / 1,180,000 |
0.00017% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
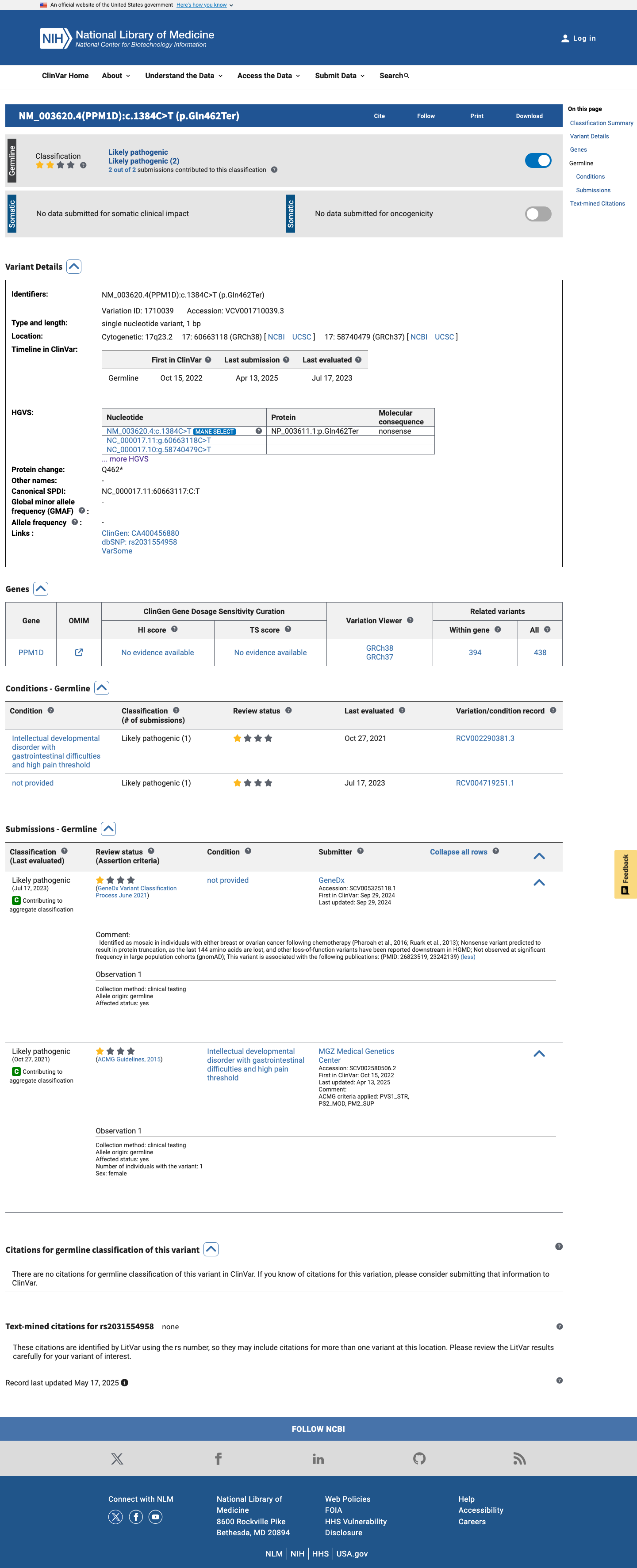
ClinVar
This variant has been reported in ClinVar as Likely pathogenic (2 clinical laboratories). (ClinVarID = 1710039)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). BayesDel score = 0.66.
Functional
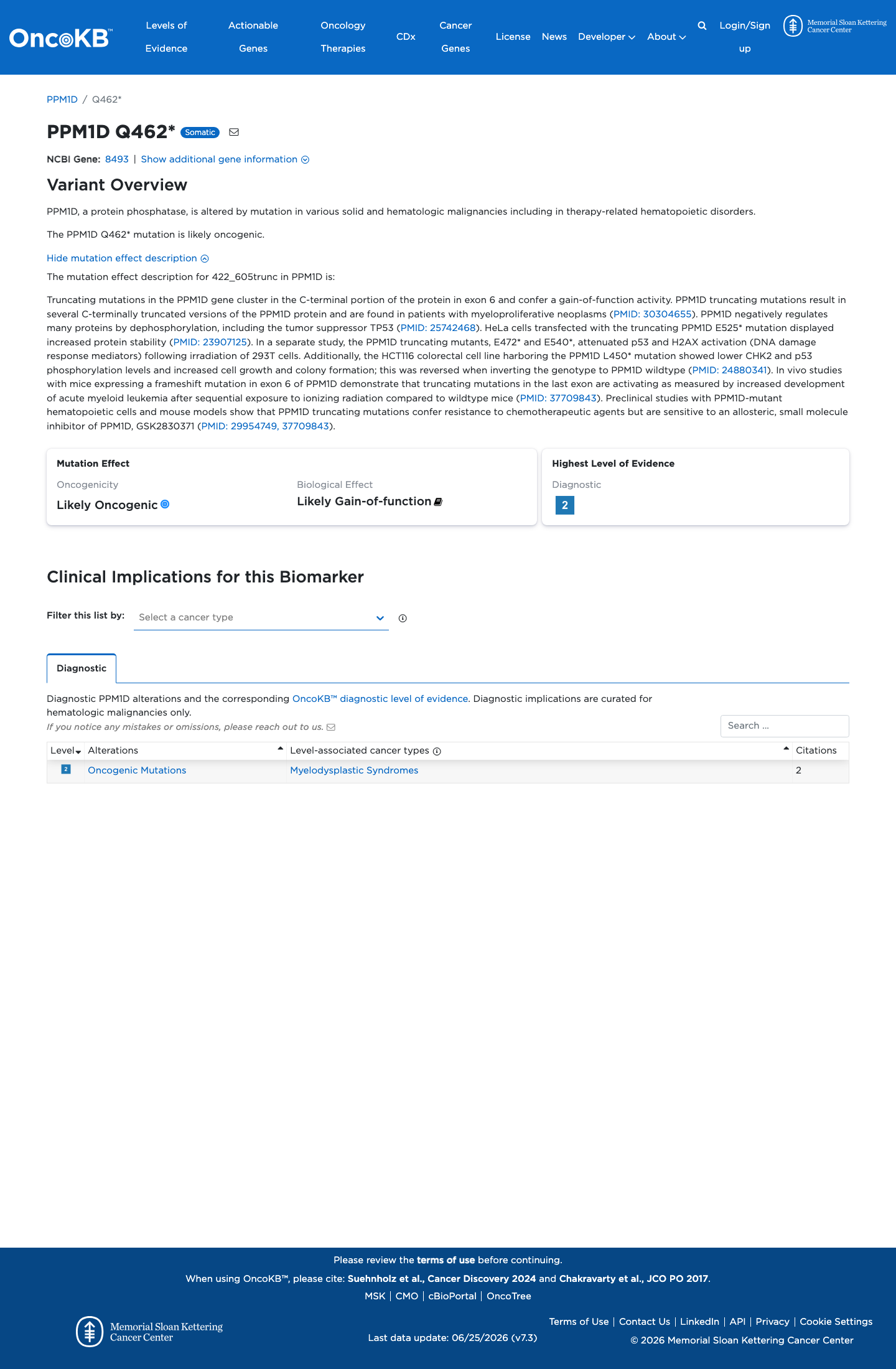
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Gain-of-function; curated oncogenicity label: Likely Oncogenic.
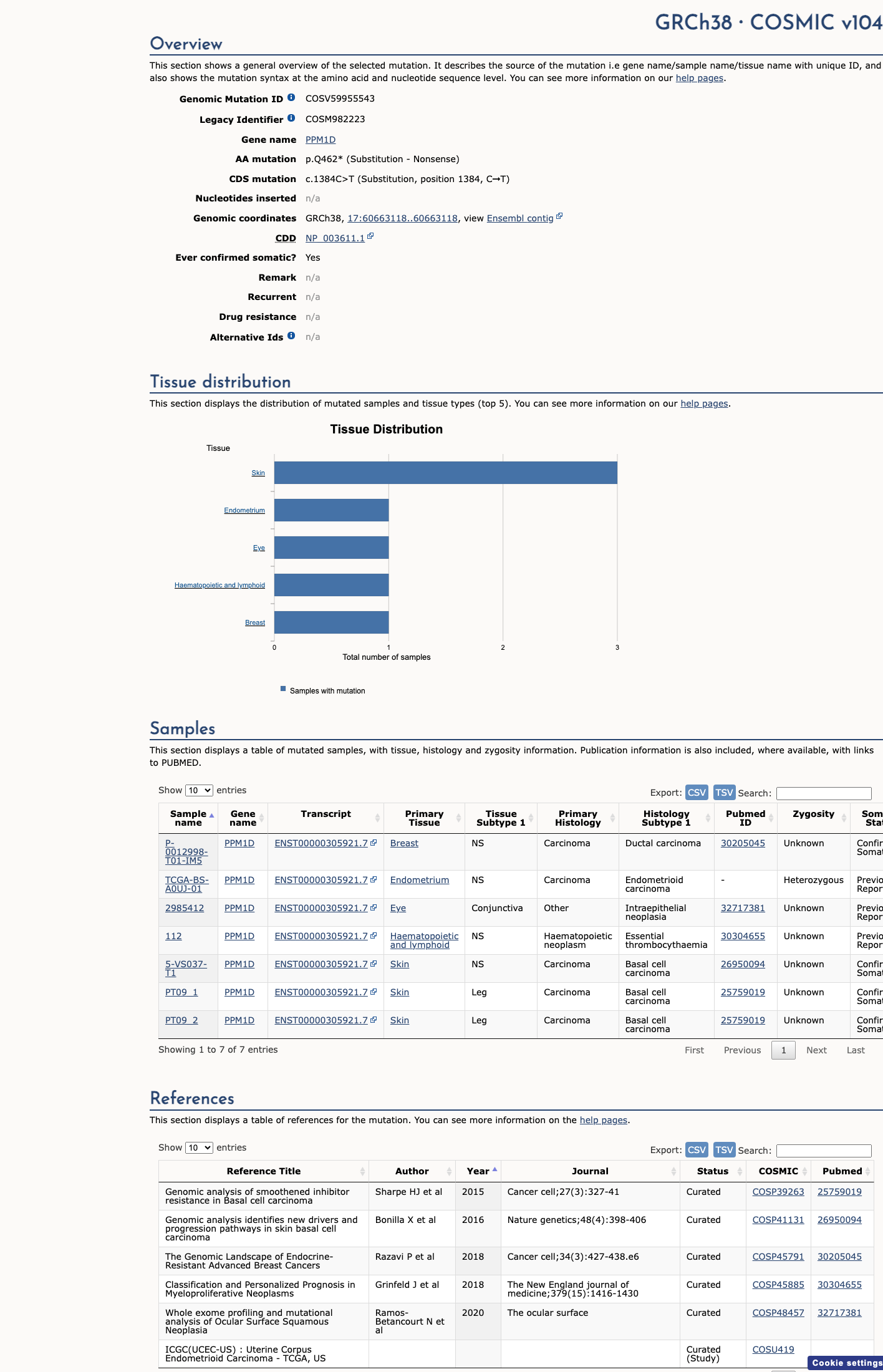
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV59955543, n = 7 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
3papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 3 further PMIDs triaged but not cited — see Sources & References.
Genetic variants and mutations of PPM1D control the response to DNA damage.
Searched
c.1384C>Tp.Gln462TerQ462XQ462*
Found
Q462X is listed among PPM1D somatic mutations identified in primary human uterine corpus endometrioid carcinoma. The paper characterizes truncating mutations in the PPM1D C-terminus as gain-of-function (E525X) or loss-of-function (R552X) depending on position and protein stability, but does not provide variant-specific functional data for Q462X.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
Why
Variant listed among cancer-associated PPM1D mutations in a mutation table; supports PM1 (hotspot region) but no variant-specific functional characterization.
Q462X
Location Table 2, Uterine corpus endometrioid carcinoma mutations list · full text
Truncating mutations of PPM1D are found in blood DNA samples of lung cancer patients.
Searched
c.1384C>Tp.Gln462TerQ462XQ462*1384
Found
Study of PPM1D truncating mutations in blood DNA of 543 non-small cell lung cancer patients. The gain-of-function nature of c.1384C>T (p.Q462X) is reported as having been assessed and confirmed by prior functional studies (Ruark et al. 2013). The paper states that all truncating mutations in the exon 6 mutation cluster region of PPM1D are assumed to be gain-of-function.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
→PS3 supports · met
Why
Variant-specific functional confirmation reported via secondary citation; referenced in PS3 assessment at supporting level and PM1 for hotspot localization.
The gain-of-function nature of the following mutations was assessed and confirmed: c.1384C>T (p.Q462X), c.1420delC (Ruark), c.1349delT (p.450X), c.1372C>T (p.R458X) (Kleiblova). It is assumed that all truncating mutations in the mutation cluster region of PPM1D are of gain-of-function type.
Location Discussion, paragraph on mutational consequences · Context Functional confirmation cited from prior studies (Ruark et al. 2013; Kleiblova et al. 2013) · full text
Classification and Personalized Prognosis in Myeloproliferative Neoplasms.
Searched
c.1384C>Tp.Gln462TerQ462XQ462*
Found
Genomic classification study of 2035 patients with myeloproliferative neoplasms. PPM1D Q462* was identified among truncating mutations in exon 6, found in the myeloproliferative neoplasm clone of a patient with essential thrombocythemia (PD6634). PPM1D was the eighth most commonly mutated gene in the cohort.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
Why
Variant directly identified in a patient with essential thrombocythemia; supports PM1 as evidence of the variant occurring within the recognized mutational hotspot.
Q462*
Location Figure 1B (PPM1D mutations map) and Figure 1C (clonal structure) · full text
Sources & reference links
Triaged references · 3 PMIDs not cited in assessment
24880341 ↗
Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas.
ONCOKB
29954749 ↗
PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells.
ONCOKB
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR