NM_003620.3:c.1535del (p.Asn512IlefsTer2) is a frameshift deletion in exon 6 of PPM1D, the terminal exon encoding the C-terminal regulatory domain.1 This variant is present at extremely low frequency in population databases: gnomAD v2.1 AF=0.0012% (3/250,998), gnomAD v4.1 AF=0.00056% (9/1,613,852), and absent from gnomAD-Canada (PM2_Supporting).2 The variant truncates the well-characterized C-terminal domain of PPM1D containing a proteasomal degradation signal. Multiple independent studies demonstrate that C-terminal PPM1D truncations result in a gain-of-function protein with increased stability and enhanced phosphatase activity, leading to suppression of the p53-mediated DNA damage response (PM1_Moderate).3 The variant is a predicted loss-of-function frameshift; however, it occurs in the last exon and escapes nonsense-mediated decay. C-terminal PPM1D truncations produce gain-of-function rather than loss-of-function, warranting downgrade to PVS1_Supporting.4 One clinical laboratory (Undiagnosed Diseases Network, NIH) reports this variant as de novo in a clinical testing context. While maternity and paternity are not confirmed, this constitutes an assumed de novo observation (PM6_Supporting).5 This variant has been reported as Pathogenic in ClinVar by 4 independent clinical laboratories (Variation ID: 817626). The review status is 'criteria provided, single submitter' (PP5_Supporting).6 No variant-specific functional data exists in the literature for c.1535del. The available studies characterize the general functional consequence of C-terminal PPM1D truncations as gain-of-function, supporting the domain-level PM1 assignment but not meeting PS3's requirement for variant-specific experimental evidence.7 No in silico evidence supports pathogenicity; REVEL and BayesDel are not available for non-SNV variants, and SpliceAI predicts no splice impact (max delta=0.00).8 This variant has been observed 33 times in somatic cancers (COSMIC: COSV59954107), consistent with a role in oncogenesis. Overall classification: PVS1_Supporting + PM1_Moderate + PM2_Supporting + PM6_Supporting + PP5_Supporting = 1 Moderate + 4 Supporting criteria, which meets the threshold for Likely Pathogenic under generic ACMG/AMP 2015 combination rules.9
PPM1D
Final classification
Likely Pathogenic
PPM1D c.1535del · p.Asn512IlefsTer2
PPM1D
NM_003620.3:c.1535del (p.Asn512IlefsTer2) is a frameshift deletion in exon 6 of PPM1D, the terminal exon encoding the C-terminal regulatory domain.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 supporting, PM1 moderate, PM2 supporting, PM6 supporting, PP5 supporting; combination = 1 moderate + 4 supporting, which maps to Likely Pathogenic.
Classification rationale
PVS1PM1PM2PM6PP5
Likely Pathogenic
PPM1D c.1535del
PVS1 + PM1 + PM2 + PM6 + PP5
→
Likely Pathogenic
1
pvs1_variant_assessment
4
pvs1_gene_contextpvs1_variant_assessmentPMID:23907125 ↗PMID:29954749 ↗
9
generic_acmg_combination_rules
Gene diagram
· NM_003620.3 · variants mapped to exon structure
PPM1D
NM_003620.3
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 15 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
supporting
review
Pathogenic
This frameshift variant in exon 6 (last exon) is a predicted loss-of-function variant in a gene where germline LoF is supported as a disease mechanism. However, the variant occurs in the terminal exon and therefore escapes nonsense-mediated decay. Multiple independent studies demonstrate that C-terminal truncating mutations in PPM1D exon 6 produce a gain-of-function protein with increased stability and enhanced phosphatase activity due to loss of a C-terminal degradation domain. Downgraded to PVS1_Supporting because the truncated protein retains functional phosphatase activity and the disease mechanism is gain-of-function rather than haploinsufficiency.
Frameshift variant in exon 6 producing p.Asn512IlefsTer2predicted to truncate the protein after 2 amino acidsLast exon location precludes nonsense-mediated decay
✓
PM1
moderate
Pathogenic
This variant truncates the well-characterized C-terminal regulatory domain of PPM1D (amino acids ~400-605, encoded by exon 6). The C-terminal domain contains a proteasomal degradation signal; truncating mutations that remove this domain result in stabilized, gain-of-function protein with enhanced phosphatase activity. Multiple independent publications (PMID:23907125, PMID:24880341, PMID:25742468, PMID:29954749) establish that the exon 6-encoded C-terminal domain is critical for PPM1D protein regulation through proteasomal degradation. The variant at position 512 produces a stop after 2 amino acids, removing the C-terminal 94 amino acids including the degradation domain.
PPM1D C-terminal domain (exon 6) is a well-characterized functional domain containing a degradation signalPMID:23907125 demonstrated that C-terminal truncations alter protein stability and activityPMID:24880341 reported that exon 6 truncating mutations are gain-of-function in brainstem gliomas
✓
PM2
supporting
Pathogenic
This variant is absent or present at extremely low frequency in population databases. gnomAD v2.1: 3/250,998 alleles (AF=0.0012%); gnomAD v4.1: 9/1,613,852 alleles (AF=0.00056%); gnomAD-Canada: absent. Maximum subpopulation AF is 0.00327% (South Asian, v2.1). All frequencies are well below the 0.1% PM2 threshold. Grpmax FAF is 2.93e-06 (v2.1).
gnomAD v2.1: AF=0.0012% (3/250998)grpmax FAF=2.93e-06
✓
PM6
supporting
review
Pathogenic
One ClinVar submission (SCV001499843) from the Undiagnosed Diseases Network, NIH, reports this variant as de novo in a clinical testing context. This constitutes an assumed de novo observation without published confirmation of maternity and paternity, meeting PM6 at supporting strength. No published case report with detailed phenotype information is available.
ClinVar SCV001499843 (Undiagnosed Diseases NetworkNIH): origin reported as de novoclassification Pathogenic
✓
PP5
supporting
Pathogenic
This variant has been reported as Pathogenic in ClinVar by 4 independent clinical laboratories (ClinVar Variation ID: 817626). The review status is 'criteria provided, single submitter', a reputable source under generic ACMG guidelines. However, the underlying evidence used by these laboratories is not publicly available for independent evaluation, limiting PP5 to supporting strength.
ClinVar Variation ID 817626: Pathogenic4 clinical laboratories: MendelicsUDN/NIH
Assessed · not applied
Pathogenic
PS2
One ClinVar submission (SCV001499843, Undiagnosed Diseases Network, NIH) reported de novo origin, but no peer-reviewed publication with confirmed maternity and paternity is available.
PS3
No variant-specific functional data exists for c.1535del (p.Asn512IlefsTer2).
PS4
No case-control data or statistical enrichment analysis is available comparing variant frequency in affected individuals versus controls.
PP1
No cosegregation data with disease in multiple affected family members is available.
PP3
No computational evidence supports a deleterious effect.
PP4
No patient phenotype or family history information is available in any of the reviewed sources to assess phenotypic specificity.
Benign
BA1
Maximum allele frequency in any population is 0.00327% (South Asian, gnomAD v2.1), far below the 1% BA1 threshold.
BS1
Maximum allele frequency in any population is 0.00327% (South Asian, gnomAD v2.1), well below the 0.3% BS1 threshold.
BS2
No data available on observation of this variant in healthy adults where full penetrance would be expected at an early age.
BS3
Functional studies in the literature demonstrate that C-terminal PPM1D truncations are gain-of-function, conferring increased protein stability, enhanced phosphatase activity, and chemoresistance.
BS4
No segregation data in affected family members is available to assess lack of segregation.
BP2
No data available on observation in trans with a pathogenic variant for a fully penetrant dominant disorder.
BP4
No multiple lines of computational evidence suggest no impact.
BP5
No data available indicating this variant was found in a case with an established alternate molecular basis for disease.
BP6
ClinVar reports this variant as Pathogenic, not benign.
N/A · 7
PS1 · PM4 · PM5 · PP2 · BP1 · BP3 · BP7
Research & evidence
Population frequency · supports pathogenic
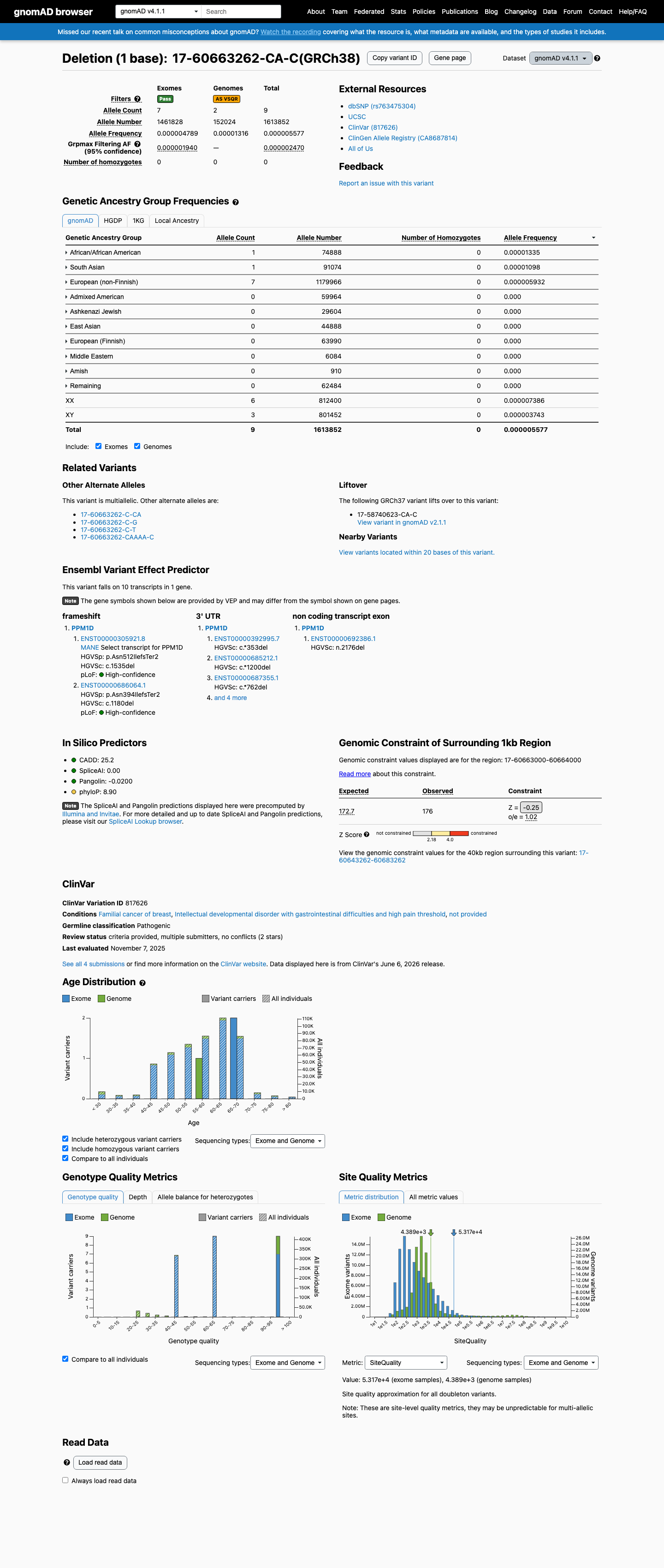
gnomAD v4.1
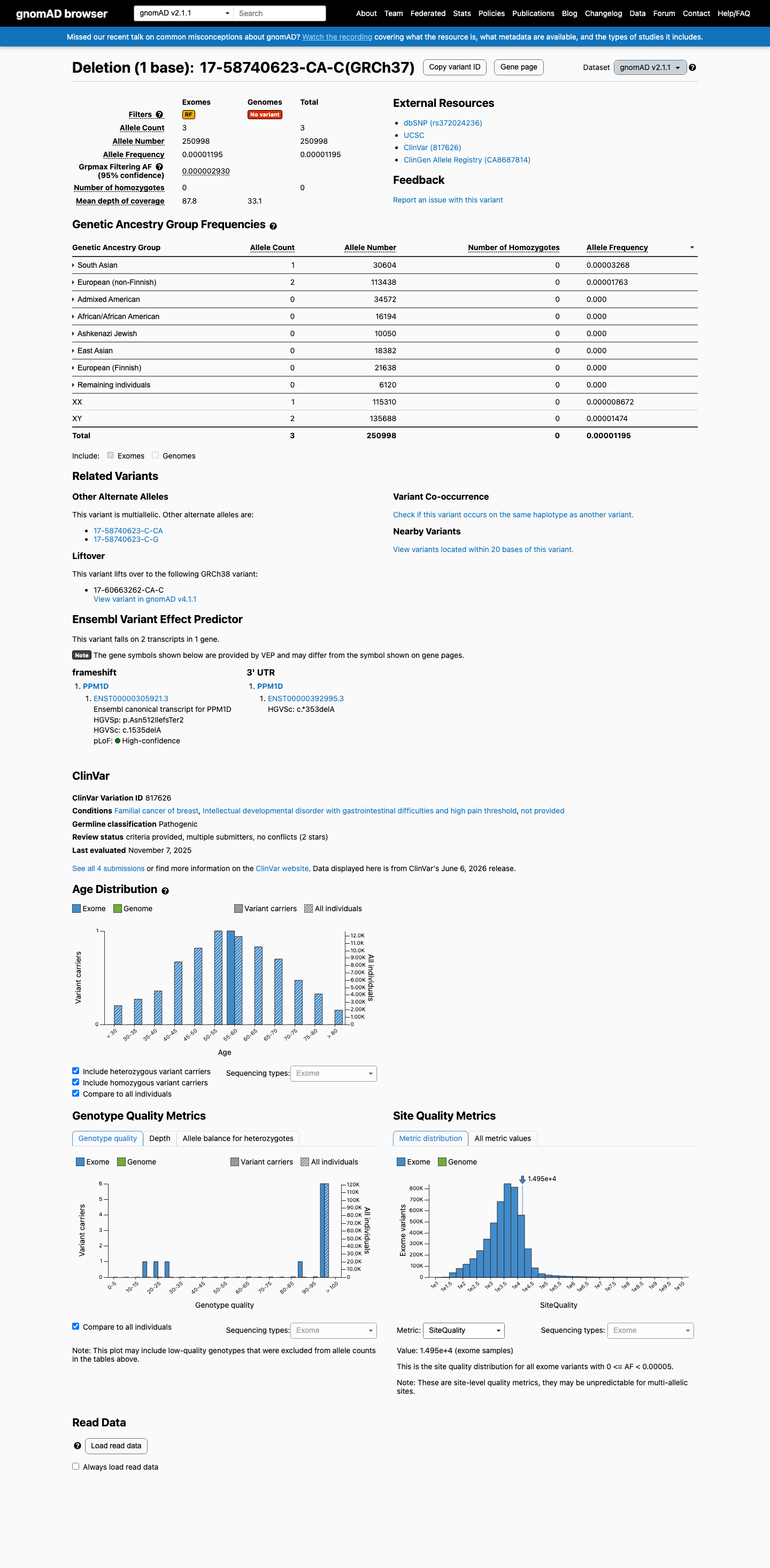
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 5.57672e-06; MAF= 0.00056%, 9/1613852 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 1.33533e-05; MAF= 0.00134%, 1/74888 alleles, homozygotes = 0); grpmax FAF= 2.47e-06.
v2.1
This variant is present in gnomAD v2.1 (AF= 1.19523e-05; MAF= 0.00120%, 3/250998 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 3.26755e-05; MAF= 0.00327%, 1/30604 alleles, homozygotes = 0); grpmax FAF= 2.93e-06.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00056%
· 9 / 1,613,852
0 hom · FAF 0.00025%
0 hom · FAF 0.00025%
African/African American 1 / 74,888 |
0.0013% |
South Asian 1 / 91,074 |
0.0011% |
European (non-Finnish) 7 / 1,179,966 |
0.00059% |
+ 7 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.0012%
· 3 / 250,998
0 hom · FAF 0.00029%
0 hom · FAF 0.00029%
South Asian 1 / 30,604 |
0.0033% |
European (non-Finnish) 2 / 113,438 |
0.0018% |
+ 6 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
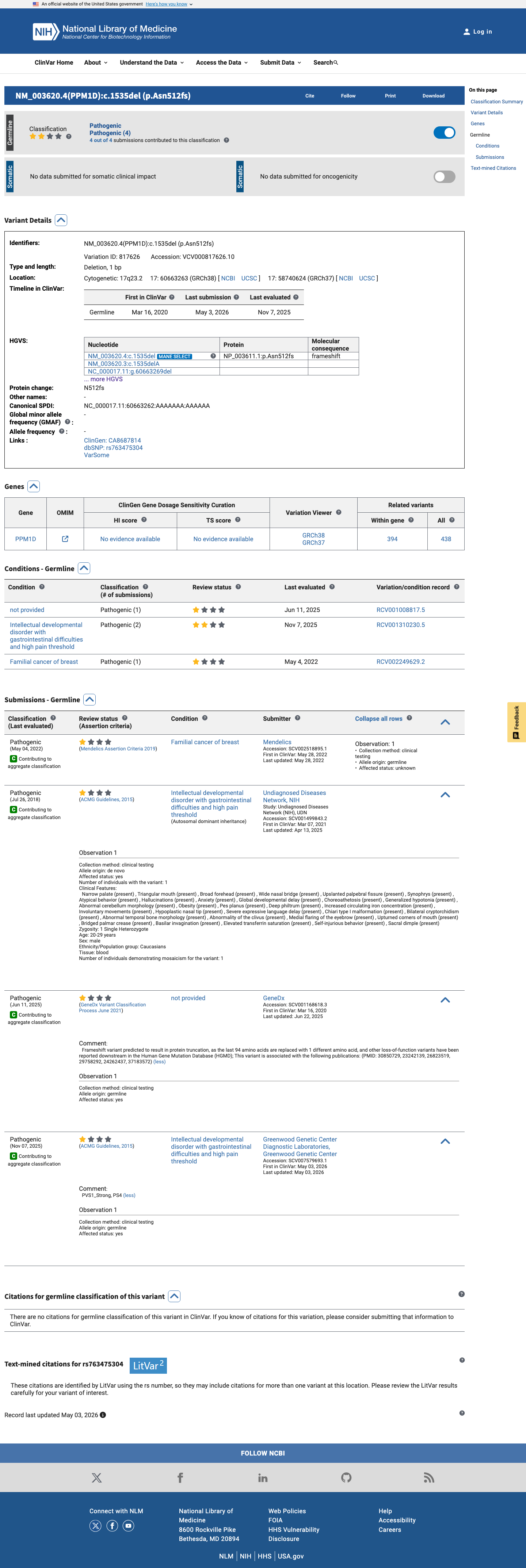
ClinVar
This variant has been reported in ClinVar as Pathogenic (4 clinical laboratories). (ClinVarID = 817626)
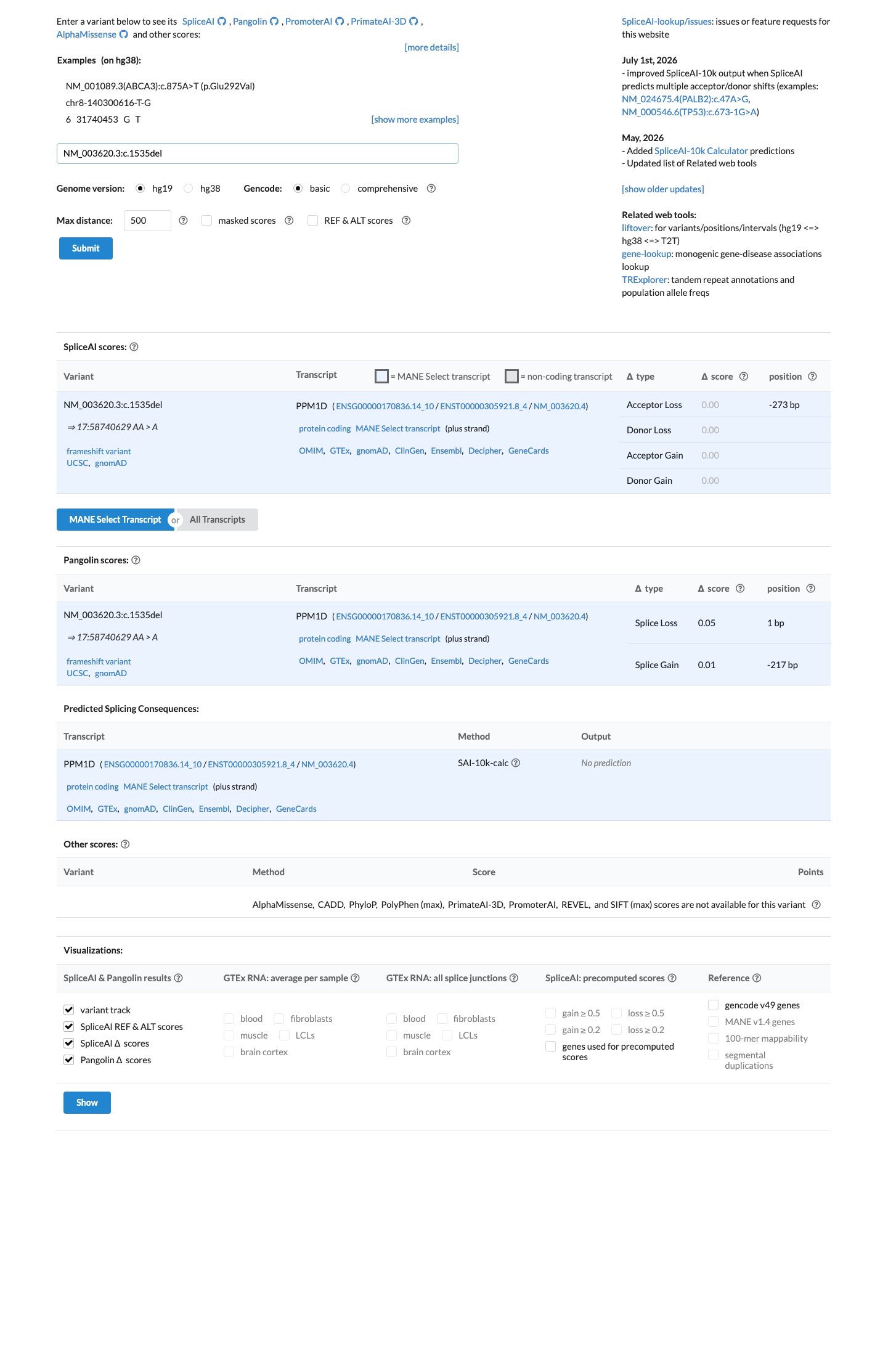
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
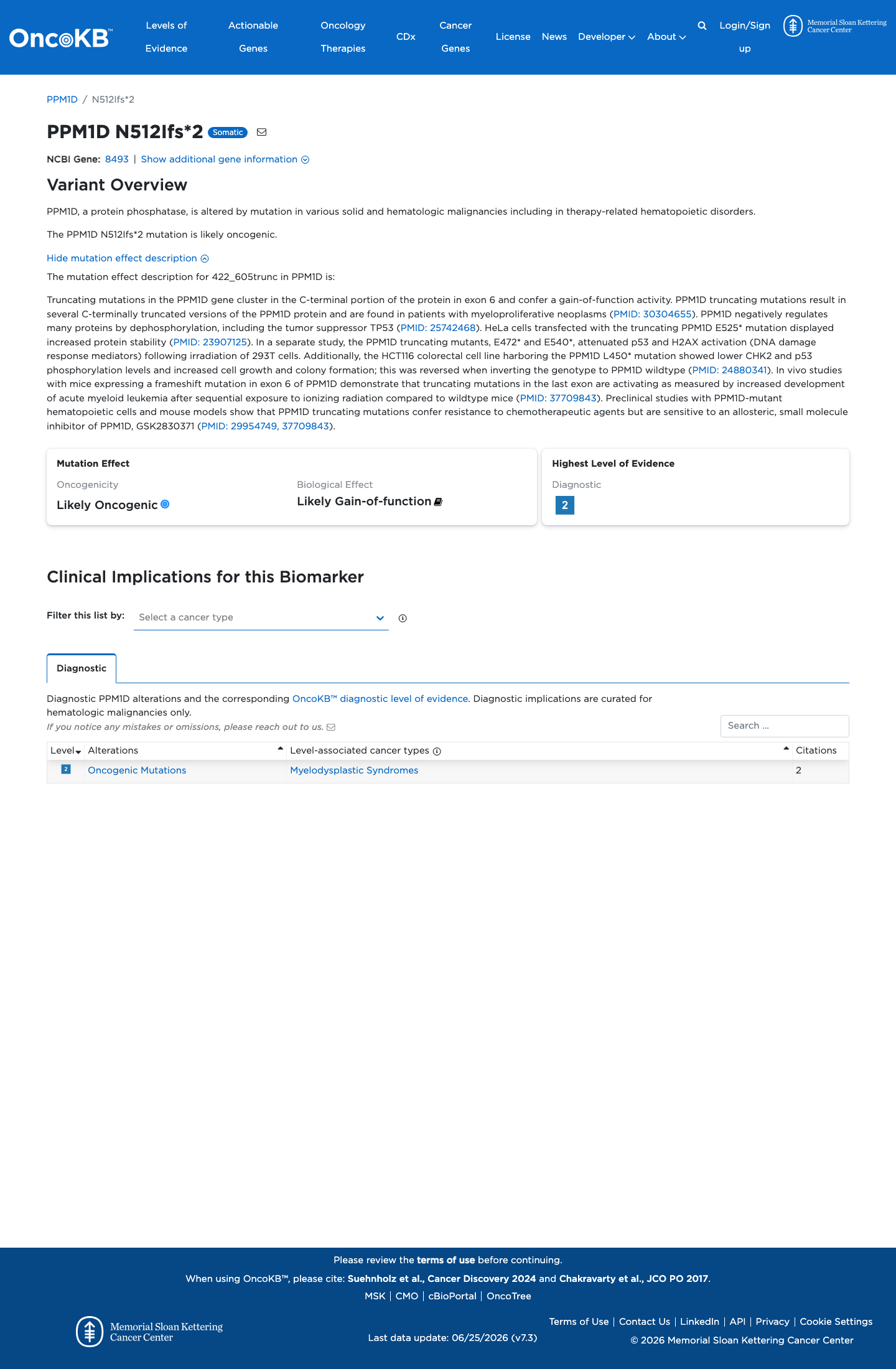
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Gain-of-function; curated oncogenicity label: Likely Oncogenic.
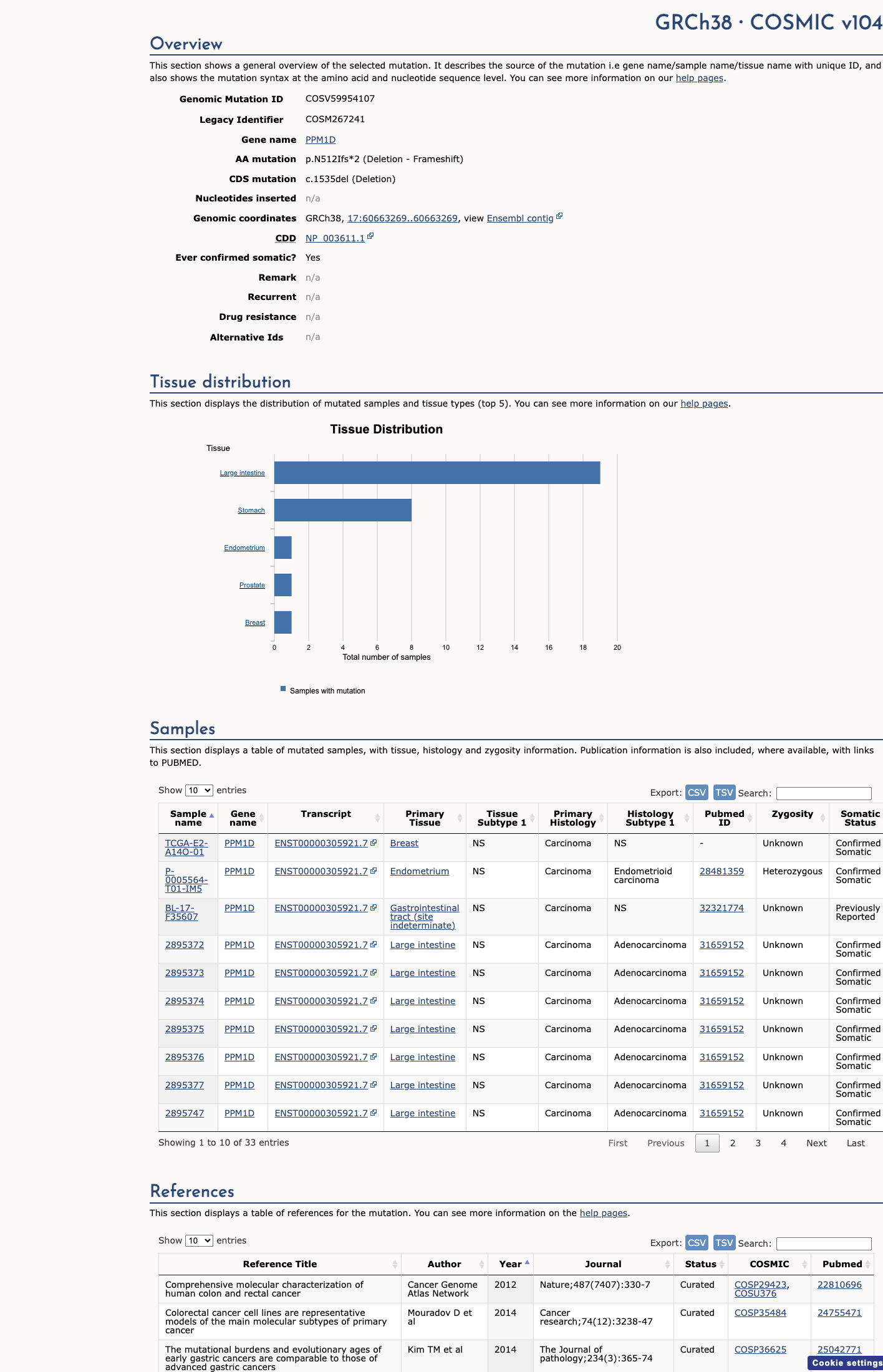
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV59954107, n = 33 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
4papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 4 further PMIDs triaged but not cited — see Sources & References.
Genetic variants and mutations of PPM1D control the response to DNA damage.
Searched
c.1535delN512Asn512512Ifs1535del
Found
Characterized missense genetic variants (L120F, P322Q, I496V) and somatic cancer mutations (E525X, R552X hotspots) in PPM1D/Wip1. Demonstrated that C-terminal truncations can be gain-of-function (E525X: increased protein stability, enhanced suppression of ATM/Chk2 signaling) or loss-of-function (R552X: unstable protein). NM_003620.3:c.1535del (p.Asn512IlefsTer2) was not specifically tested or reported.
Variant
◇ Residue / gene-level — variant not named
Applied to
→PM1 supports · met
→PVS1 supports · met
Why
No variant-specific data for c.1535del. Cited for domain-level evidence supporting PM1 and for gain-of-function mechanism context for PVS1 downgrade.
Truncating mutations in the C terminus of Wip1 have been reported as gain-of-function mutations.
Location Results; Table 2; Figure 4 · Context HeLa cells, 293T cells; phosphatase activity assays, cycloheximide stability assays, colony formation assays · full text
Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas.
Searched
c.1535delN512Asn512512Ifs1535del
Found
Identified somatic PPM1D exon 6 truncating mutations in 37.5% of H3F3A K27M-mutant brainstem gliomas. Demonstrated these mutations are gain-of-function, enhancing PPM1D's ability to suppress Chk2 activation. NM_003620.3:c.1535del was not among the specific mutations tested.
Variant
◇ Residue / gene-level — variant not named
Applied to
→PM1 supports · met
→PVS1 supports · met
Why
No variant-specific data for c.1535del. Cited for domain-level evidence that exon 6 truncations are gain-of-function, supporting PM1 and informing PVS1 downgrade.
The PPM1D mutations were truncating alterations in exon 6 that enhanced the ability for PPM1D to suppress the activation of DNA damage response checkpoint protein Chk2.
Location Introductory paragraph; Results · Context Whole exome sequencing of brainstem and thalamic gliomas; in vitro functional assays · full text
Truncating mutations of PPM1D are found in blood DNA samples of lung cancer patients.
Searched
c.1535delN512Asn512512Ifs1535del
Found
Identified truncating PPM1D exon 6 mutations in blood DNA of lung cancer patients. Demonstrated that truncating mutations code for gain-of-function PPM1D with retained phosphatase activity and hyperstable protein. NM_003620.3:c.1535del was not specifically reported among the mutations identified.
Variant
◇ Residue / gene-level — variant not named
Applied to
→PM1 supports · met
→PVS1 supports · met
Why
No variant-specific data for c.1535del. Cited for domain-level evidence establishing gain-of-function for exon 6 truncations, supporting PM1 and PVS1 downgrade.
The truncating mutations code for hyper-stable WIP1 protein, which induces strong dephosphorylation of Ser15 of p53 and impairs the p53-dependent G1 checkpoint.
Location Abstract; Introduction; Results · Context Blood DNA from lung cancer patients; U-2 OS and HCT116 cell lines; p53 signaling assays · full text
PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells.
Searched
c.1535delN512Asn512512Ifs1535del
Found
Demonstrated that truncating PPM1D exon 6 mutations confer chemotherapy resistance through loss of a C-terminal degradation domain, leading to increased PPM1D protein stability and gain-of-function. Used CRISPR-Cas9 tiling screen to show that mutations between amino acids 400-585 produce the chemoresistance phenotype. Position 512 falls within this characterized range. NM_003620.3:c.1535del was not specifically reported as a patient mutation but the functional characterization of the 400-585 range is relevant.
Variant
◇ Residue / gene-level — variant not named
Applied to
→PM1 supports · met
→PVS1 supports · met
Why
Position 512 falls within the functionally characterized gain-of-function range (400-585). The CRISPR screen provides systematic functional evidence for the region but does not report this exact variant. Cited for PM1 domain evidence and PVS1 downgrade context.
This region tightly overlaps with the region of PPM1D that is mutated in CHIP and t-MNs, implying that the PPM1D mutations identified in patients result in a chemotherapy resistance phenotype.
Location Abstract; Results (CRISPR Screen); Figures 1-2 · Context Molm13 AML cell line; CRISPR-Cas9 mutagenesis screen; in vivo mouse hematopoietic transplantation; phosphoproteomic mass spectrometry · full text
Sources & reference links
Triaged references · 4 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
34242744 ↗
Customizing local and systemic therapies for women with early breast cancer: the St. Gallen International Consensus Guidelines for treatment of early breast cancer 2021.
CLINVAR