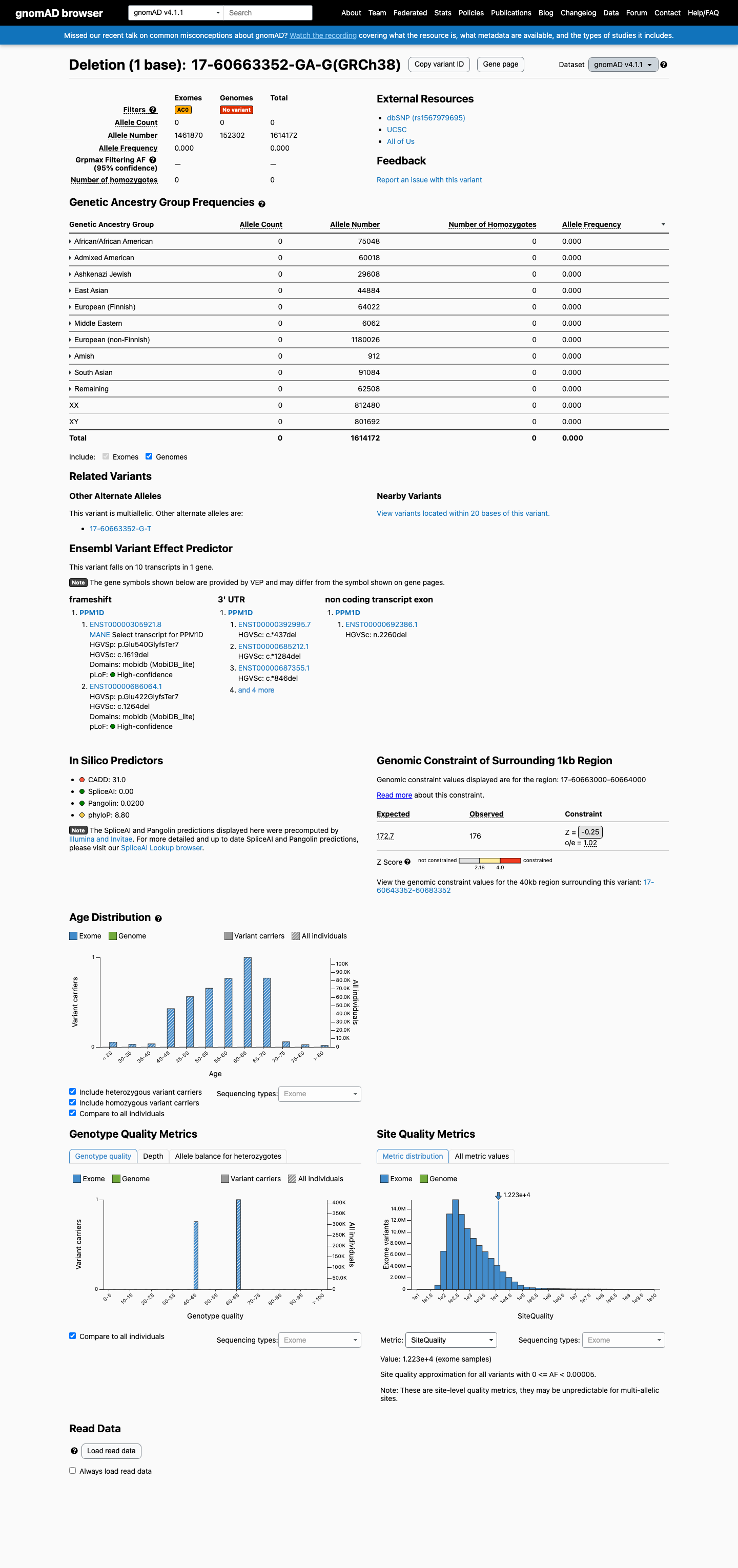
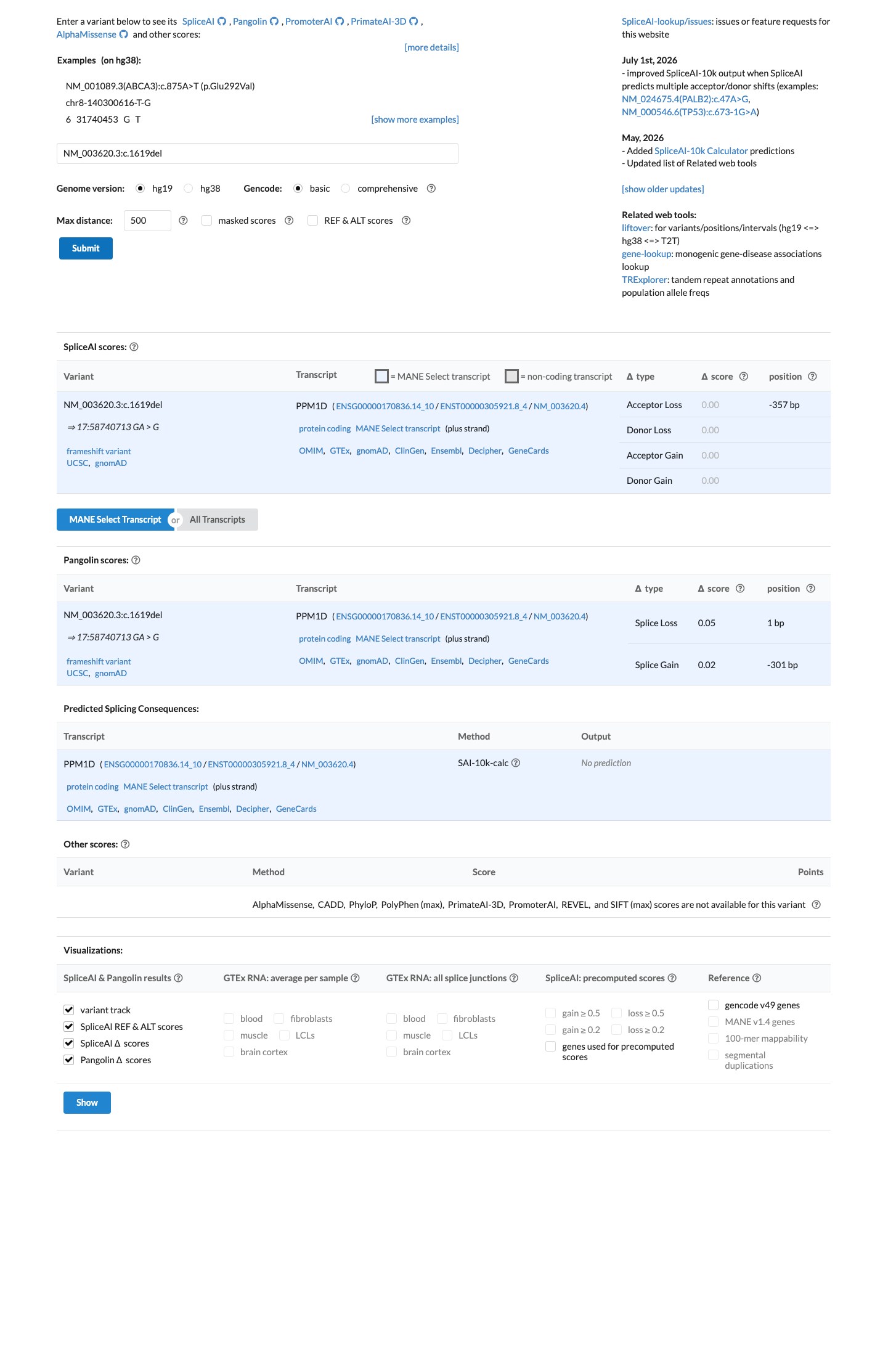
NM_003620.3:c.1619del (p.Glu540GlyfsTer7) is a frameshift deletion in exon 6 (terminal exon) of PPM1D, predicted to result in premature termination with removal of the C-terminal degradation domain.1 This variant is absent from gnomAD v2.1 and v4.1 (0/1,614,172 alleles, AF=0.0%) across all ancestral populations, and absent from gnomAD-Canada v1.0.2 The variant is absent from ClinVar and has no clinical germline classification. It has been observed as a somatic event in COSMIC (COSV99045424, n=2).3 OncoKB curates this variant as Likely Oncogenic with predicted gain-of-function effect, consistent with the broader literature on PPM1D exon 6 truncating mutations.4 Five publications were reviewed in full text; none specifically mention NM_003620.3:c.1619del (p.Glu540GlyfsTer7). PMID:24880341 reports a nonsense variant at the same codon (E540X) in brainstem gliomas in a somatic context.5 SpliceAI predicts no splicing impact (max delta score 0.00). REVEL and BayesDel scores are not available for this indel variant.6 Under the generic ACMG/AMP 2015 classification framework (PMID:25741868), PVS1_Strong and PM2_Supporting are assigned. This combination (1 Strong + 1 Supporting) does not meet the threshold for Likely Pathogenic (requires 1 Strong + 1-2 Moderate, 3 Moderate, or 2 Moderate + 2 Supporting). The overall classification is Variant of Uncertain Significance (VUS).7
PPM1D
Final classification
VUS
PPM1D c.1619del · p.Glu540GlyfsTer7
PPM1D
NM_003620.3:c.1619del (p.Glu540GlyfsTer7) is a frameshift deletion in exon 6 (terminal exon) of PPM1D, predicted to result in premature termination with removal of the C-terminal degradation domain.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 strong, PM2 supporting; combination = 1 strong + 1 supporting, which maps to VUS.
Classification rationale
PVS1PM2
VUS
PPM1D c.1619del
PVS1 + PM2
→
VUS
4
oncokb ↗
7
generic_acmg_combination_rules
Gene diagram
· NM_003620.3 · variants mapped to exon structure
PPM1D
NM_003620.3
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 17 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
strong
review
Pathogenic
NM_003620.3:c.1619del is a frameshift deletion in exon 6 (the terminal exon) of PPM1D, predicted to cause premature termination at p.Glu540GlyfsTer7, removing the C-terminal 66 amino acids including the degradation signal domain. Under ClinGen SVI PVS1 recommendations (PMC6185798), frameshift variants are eligible for PVS1 when loss of function is an established disease mechanism. PPM1D germline loss-of-function disease context is supported by literature review. However, the variant resides in the last exon and is predicted to escape nonsense-mediated decay; PPM1D exon 6 truncations have been characterized as gain-of-function in functional studies. Downgraded from PVS1_VeryStrong to PVS1_Strong per PMC6185798 due to NMD escape and residual protein expression with retained phosphatase domain, though the removed C-terminal region contains a functionally critical degradation signal.
Frameshift variant in terminal exon 6 of PPM1DPredicted protein truncation p.Glu540GlyfsTer7 removes C-terminal degradation domainGermline LoF disease mechanism supported by literature (PMC6185798 framework applied)
✓
PM2
supporting
Pathogenic
NM_003620.3:c.1619del is absent from gnomAD v2.1 and v4.1 (0/1,614,172 alleles), with zero allele count across all ancestral populations. This meets the PM2 threshold for absence in population databases (allele frequency <0.1%) under the non-VCEP generic ACMG framework.
gnomAD v2.1: absentgnomAD v4.1: 0/1614
Assessed · not applied
Pathogenic
PS2
No de novo confirmation data are available for this variant.
PS3
No variant-specific functional studies of NM_003620.3:c.1619del (p.Glu540GlyfsTer7) were identified in the reviewed literature.
PS4
No case-control studies or cohort reports documenting NM_003620.3:c.1619del in affected individuals were identified.
PM1
The variant does not lie in a statistically significant mutational hotspot as determined by cancerhotspots.org analysis.
PM6
No de novo data are available for this variant.
PP1
No segregation data are available for this variant.
PP3
In silico pathogenicity prediction tools (REVEL, BayesDel) are not applicable to indel variants and returned no scores.
PP4
No specific patient phenotype data are available for individuals harboring NM_003620.3:c.1619del as a germline variant.
Benign
BA1
NM_003620.3:c.1619del is absent from gnomAD v4.1 (0/1,614,172 alleles, AF=0.0%).
BS1
NM_003620.3:c.1619del is absent from gnomAD v4.1 (0/1,614,172 alleles, AF=0.0%).
BS2
BS2 requires observation of the variant in homozygous state or in trans with a pathogenic variant in healthy individuals.
BS3
No variant-specific functional studies demonstrate a benign or neutral effect for NM_003620.3:c.1619del.
BS4
No segregation data are available to demonstrate lack of cosegregation with disease.
BP2
BP2 requires observation of the variant in trans with a known pathogenic dominant variant, or in cis with a pathogenic variant.
BP3
BP3 applies to in-frame deletions/insertions in repetitive regions without a known function.
BP4
BP4 requires multiple lines of computational evidence suggesting no impact on gene or gene product.
BP5
BP5 requires observation of the variant in a case with an alternate molecular basis for disease.
N/A · 9
PS1 · PM3 · PM4 · PM5 · PP2 · PP5 · BP1 · BP6 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0; MAF= 0.00000%, 0/1614172 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0; MAF= 0.00000%, 0/75048 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / 1,614,172
0 hom
0 hom
Not observed in any ancestry group.
+ 10 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American, European (non-Finnish))
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
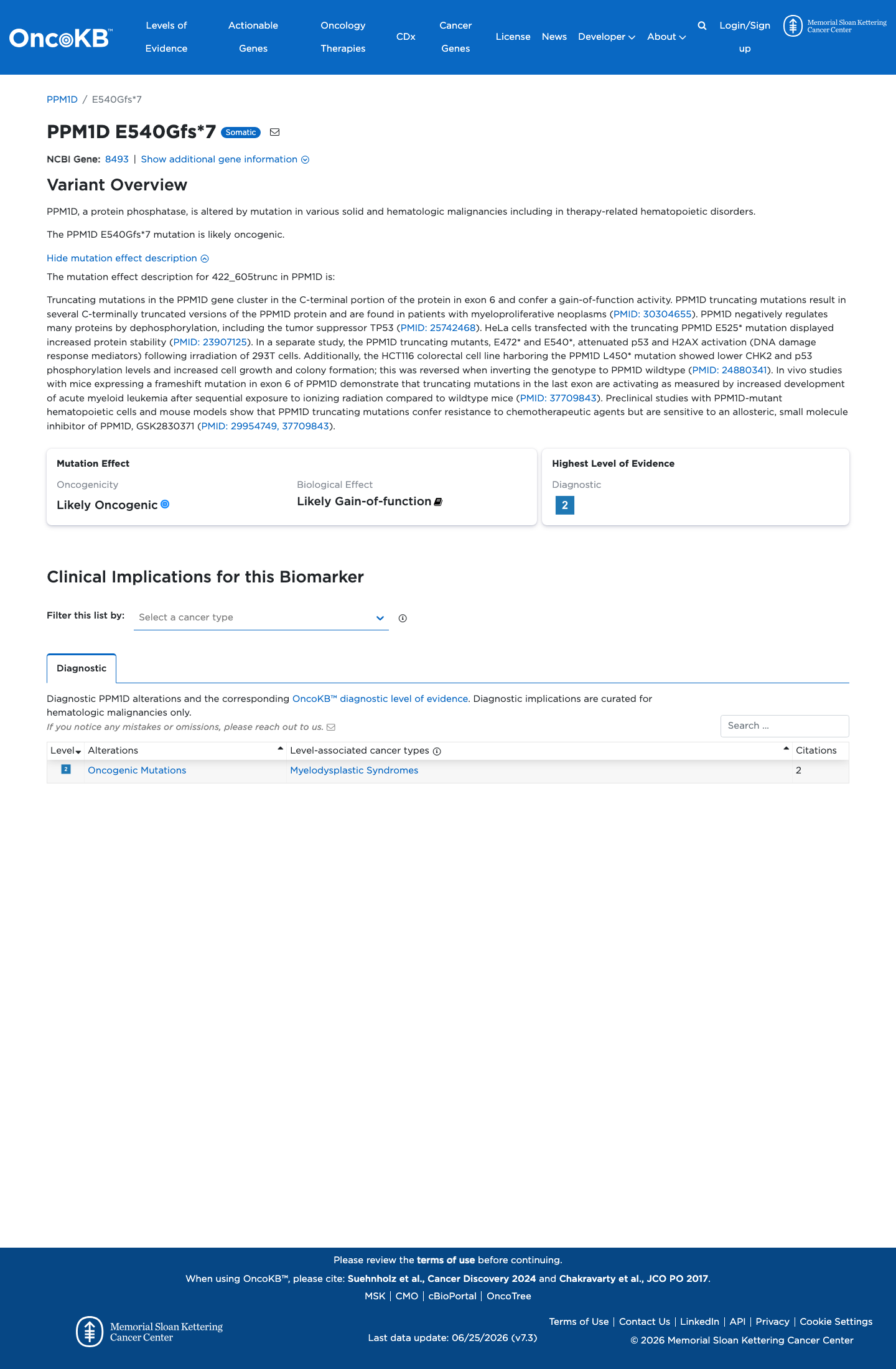
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Gain-of-function; curated oncogenicity label: Likely Oncogenic.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
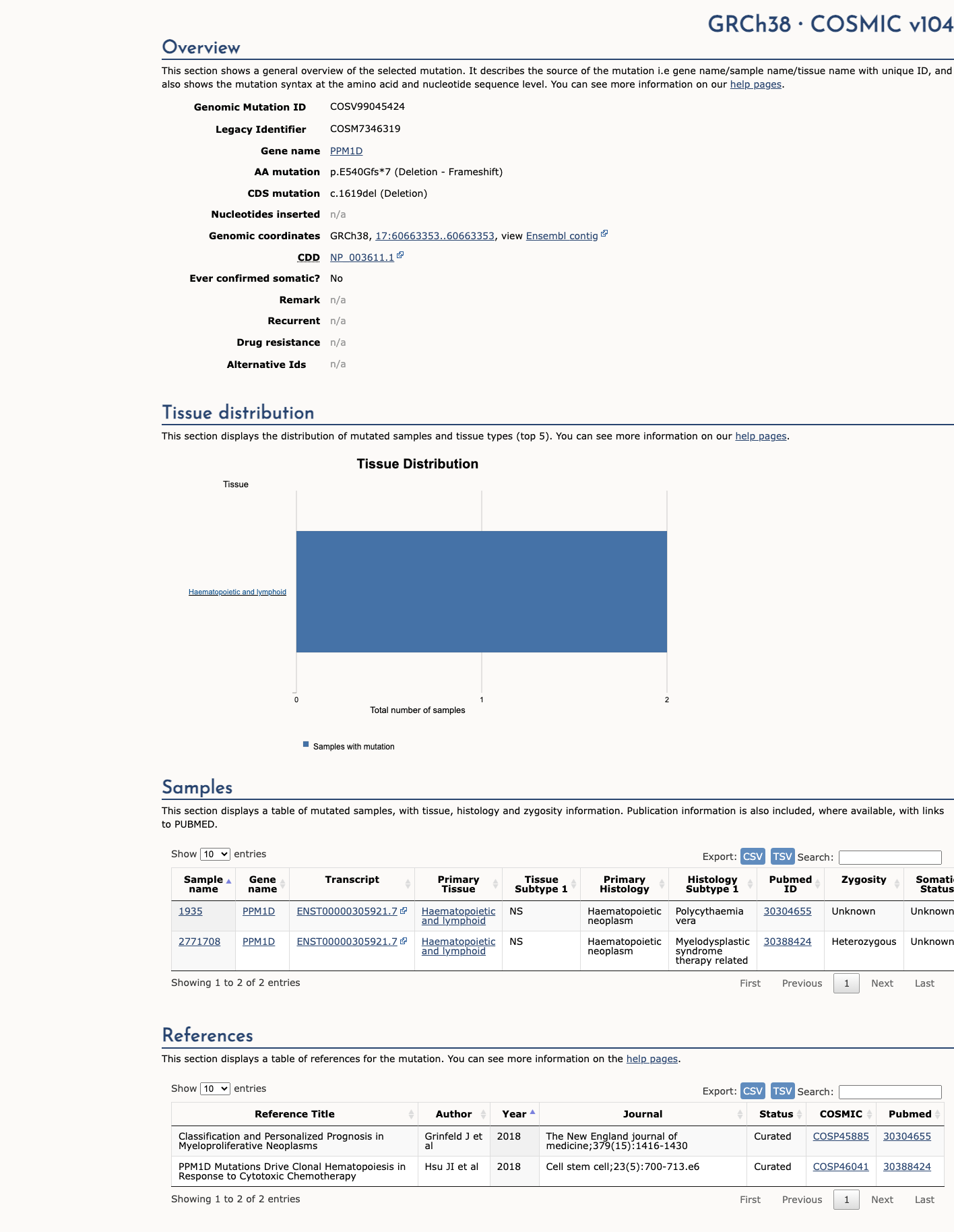
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV99045424, n = 2 times).
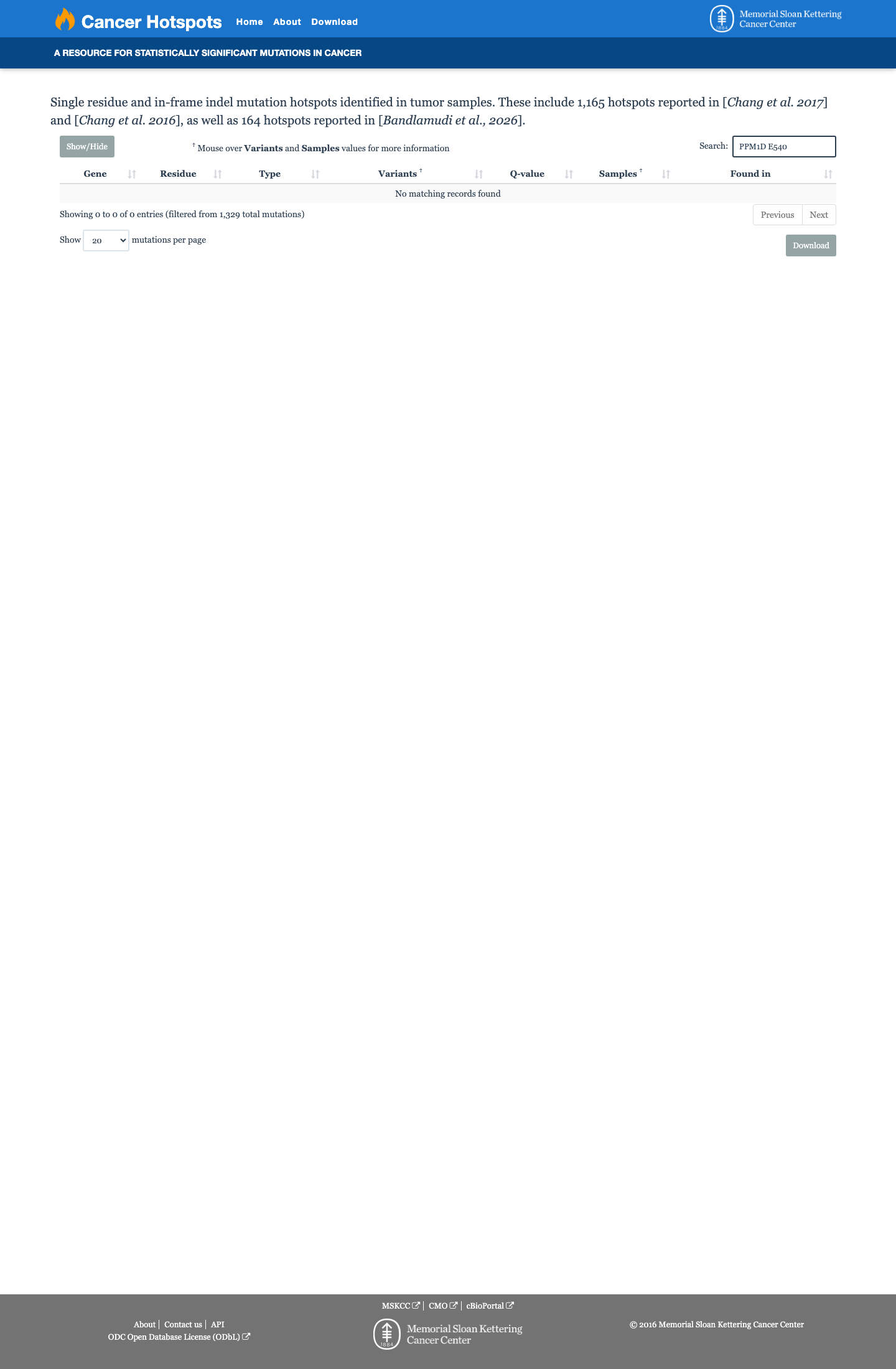
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 5 PMIDs not cited in assessment
24880341 ↗
Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas.
ONCOKB
25742468 ↗
Truncating mutations of PPM1D are found in blood DNA samples of lung cancer patients.
ONCOKB
29954749 ↗
PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells.
ONCOKB