NM_004360.5:c.2053G>A (p.Val685Met) is a missense variant in CDH1 exon 13. The ClinGen CDH1 Expert Panel has classified this variant as Likely benign (ClinVar Variation ID: 133847).1 The variant is present in gnomAD population databases at low frequency: 4/251,468 alleles in v2.1 (AF 0.00159%) and 24/1,613,720 alleles in v4.1 (AF 0.00149%), with one homozygote reported in v4.1. These frequencies exceed the CDH1 VCEP PM2 threshold of ≤1/100,000 alleles, and the homozygote observation meets BP2_Supporting under the VCEP rules.2 SpliceAI predicts no significant splicing impact (max delta score 0.11). In silico protein predictors show borderline scores (REVEL 0.47; BayesDel -0.1394), but per CDH1 VCEP, protein-based computational models are not used for PP3/BP4 assessment.3 No variant-specific functional studies, segregation data, de novo reports, or case-control studies were identified in the reviewed literature. Six full-text papers and 10 additional abstracts were reviewed; none mention NM_004360.5:c.2053G>A or p.Val685Met. One homozygote in gnomAD v4.1 meets CDH1 VCEP BP2_Supporting ('observed in the homozygous state in gnomAD'), providing a single line of benign evidence. No other ACMG criteria are met with the available evidence. Most criteria remain unassessed due to lack of variant-specific functional, segregation, or clinical phenotype data.4
CDH1
Final classification
Likely Benign
CDH1 c.2053G>A · p.Val685Met
CDH1
NM_004360.5:c.2053G>A (p.Val685Met) is a missense variant in CDH1 exon 13. The ClinGen CDH1 Expert Panel has classified this variant as Likely benign (ClinVar Variation ID: 133847).
ClinGen CDH1 Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines Version 3.1 v3.1.0 lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BP2 supporting benign, BP6 supporting benign; combination = 2 supporting benign, which maps to Likely Benign.
Classification rationale
BP2BP6
Likely Benign
CDH1 c.2053G>A
BP2 + BP6
→
Likely Benign
Gene diagram
· NM_004360.5 · variants mapped to exon structure
CDH1
NM_004360.5
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 14 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
BP2
supporting
review
Benign
Per CDH1 VCEP BP2_Supporting rule: 'observed in the homozygous state in gnomAD.' gnomAD v4.1 reports 1 homozygote for this variant (total_hom = 1). This meets the VCEP-specified threshold for BP2 at the Supporting benign level. The single homozygote observation in a large population database (1,613,720 alleles) is consistent with a variant unlikely to cause a highly penetrant dominant disorder like HDGC. Note: a single homozygote in gnomAD may represent a technical artifact; the expert panel's Likely benign classification provides orthogonal support.
gnomAD v4.1: 1 homozygote (exome)ClinGen CDH1 VCEP classification: Likely benign
Assessed · not applied
Pathogenic
PS2
No de novo occurrence data identified for this variant.
PS3
Per CDH1 VCEP, PS3 requires RNA assay demonstrating abnormal transcripts.
PS4
PS4 requires families meeting HDGC criteria.
PM2
Per CDH1 VCEP, PM2_Supporting requires ≤1 out of 100,000 alleles in the gnomAD cohort.
PM6
No assumed de novo occurrences identified.
PP1
No cosegregation data identified for this variant.
PP3
Per CDH1 VCEP, PP3 is limited to splicing predictions only (do not use protein-based computational models).
Benign
BA1
Per CDH1 VCEP, BA1 requires MAF ≥0.2%.
BS1
Per CDH1 VCEP, BS1 requires MAF ≥0.1%.
BS2
BS2 requires phenotype data on individuals carrying the variant — specifically, individuals without gastric cancer, diffuse gastric cancer, signet ring cell tumors, or lobular breast cancer, and whose families do not suggest HDGC.
BS3
Per CDH1 VCEP, BS3 requires functional RNA studies demonstrating no impact on transcript composition and is limited to synonymous, intronic, or non-coding variants.
BS4
No segregation data available for this variant.
BP4
Per CDH1 VCEP, BP4 is limited to splicing predictions only and requires at least three in silico splicing predictors in agreement (SpliceAI, MaxEntScan, SSF, GeneSplicer, HSF, TraP, varSEAK).
BP5
BP5 requires identification of a pathogenic/likely pathogenic variant in an alternate gene known to cause HDGC (per VCEP: currently only CTNNA1).
N/A · 9
PVS1 · PS1 · PM1 · PM5 · PP2 · PP4 · PP5 · BP1 · BP7
Research & evidence
Population frequency · supports benign
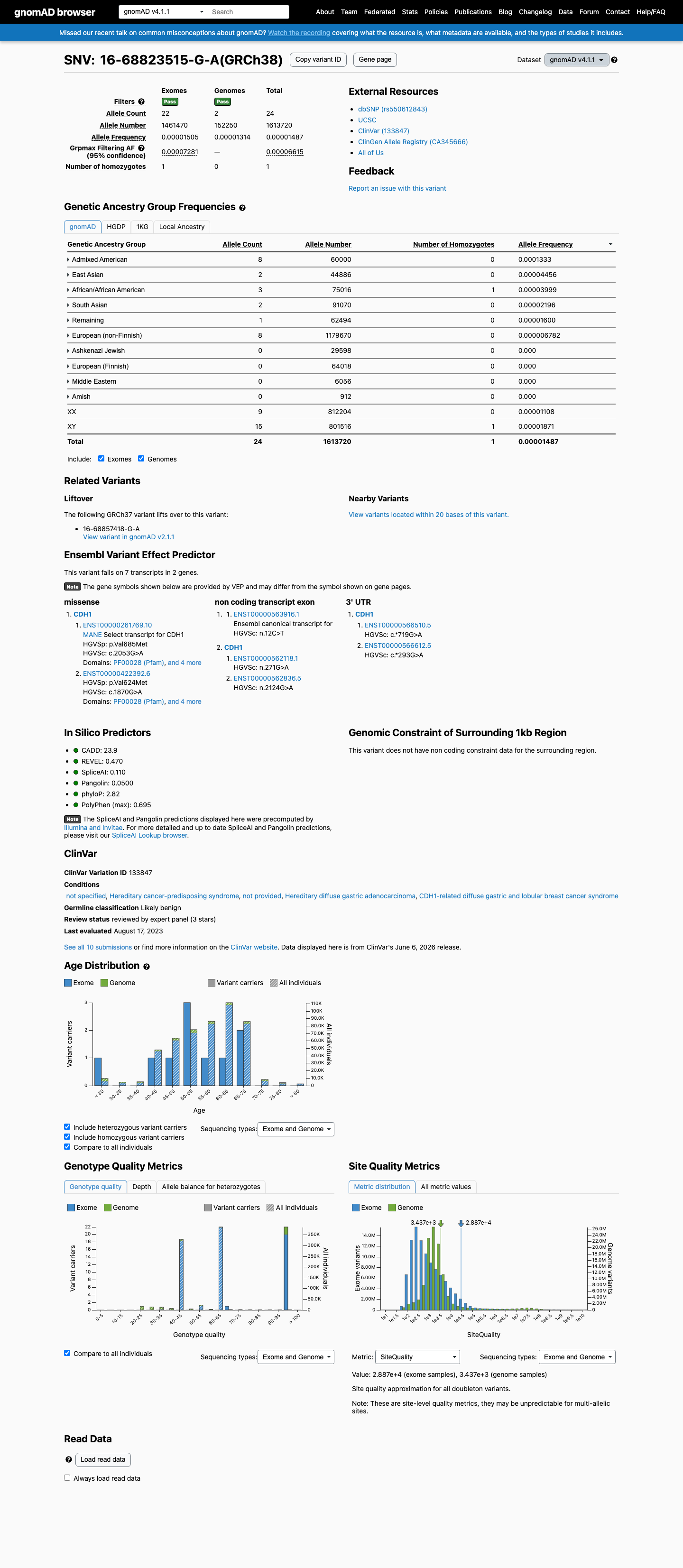
gnomAD v4.1
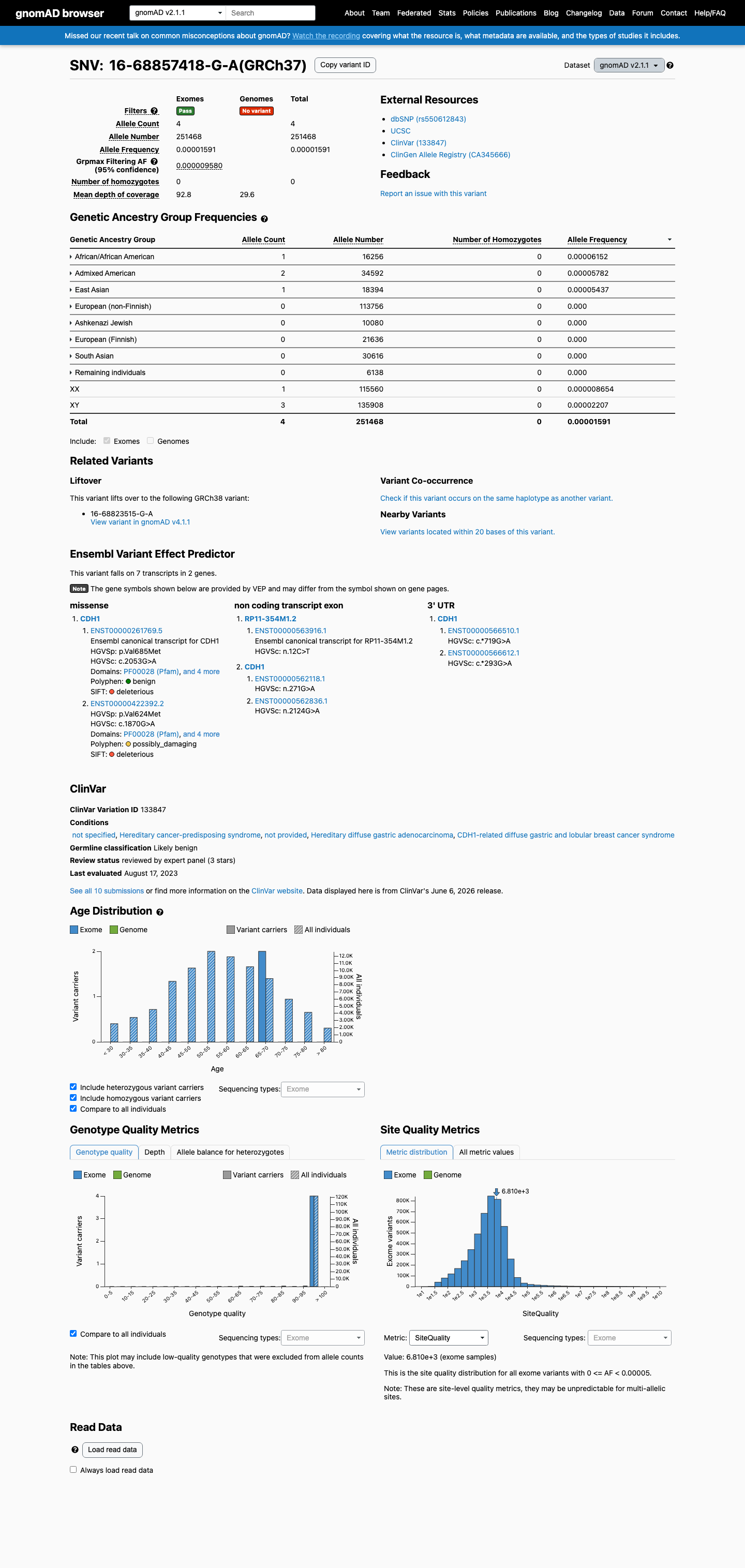
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 1.48725e-05; MAF= 0.00149%, 24/1613720 alleles, homozygotes = 1) and has highest observed frequency in the Admixed American population (AF= 0.000133333; MAF= 0.01333%, 8/60000 alleles, homozygotes = 0); grpmax FAF= 6.615e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 1.59066e-05; MAF= 0.00159%, 4/251468 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 6.15157e-05; MAF= 0.00615%, 1/16256 alleles, homozygotes = 0); grpmax FAF= 9.58e-06.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.00010856584518510477, 2/18422 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0015%
· 24 / 1,613,720
1 hom · FAF 0.0066%
1 hom · FAF 0.0066%
Admixed American 8 / 60,000 |
0.013% |
East Asian 2 / 44,886 |
0.0045% |
African/African American 3 / 75,016 |
0.004% 1 hom |
South Asian 2 / 91,070 |
0.0022% |
Remaining individuals 1 / 62,494 |
0.0016% |
European (non-Finnish) 8 / 1,179,670 |
0.00068% |
+ 4 not observed (European (Finnish), Amish, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.0016%
· 4 / 251,468
0 hom · FAF 0.00096%
0 hom · FAF 0.00096%
African/African American 1 / 16,256 |
0.0062% |
Admixed American 2 / 34,592 |
0.0058% |
East Asian 1 / 18,394 |
0.0054% |
+ 5 not observed (Ashkenazi Jewish, European (Finnish), European (non-Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
0.011%
· 2 / 18,422
0 hom · FAF 0.042%
0 hom · FAF 0.042%
Latino/Admixed American 2 / 838 |
0.24% |
+ 8 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, European (non-Finnish), Remaining individuals, South Asian)
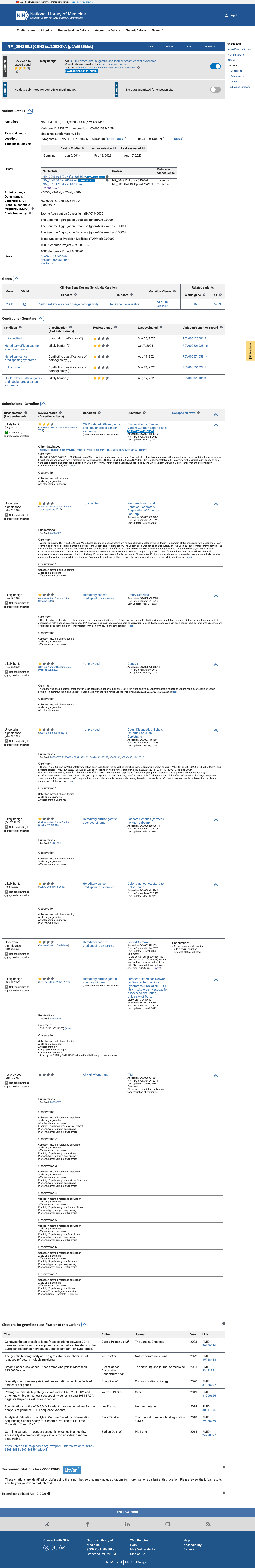
ClinVar
This variant has been reported in ClinVar as Likely benign (5 clinical laboratories) and as Uncertain significance (2 clinical laboratories) and as Likely benign by Clingen Gastric Cancer Variant Curation Expert Panel (expert panel). (ClinVarID = 133847)
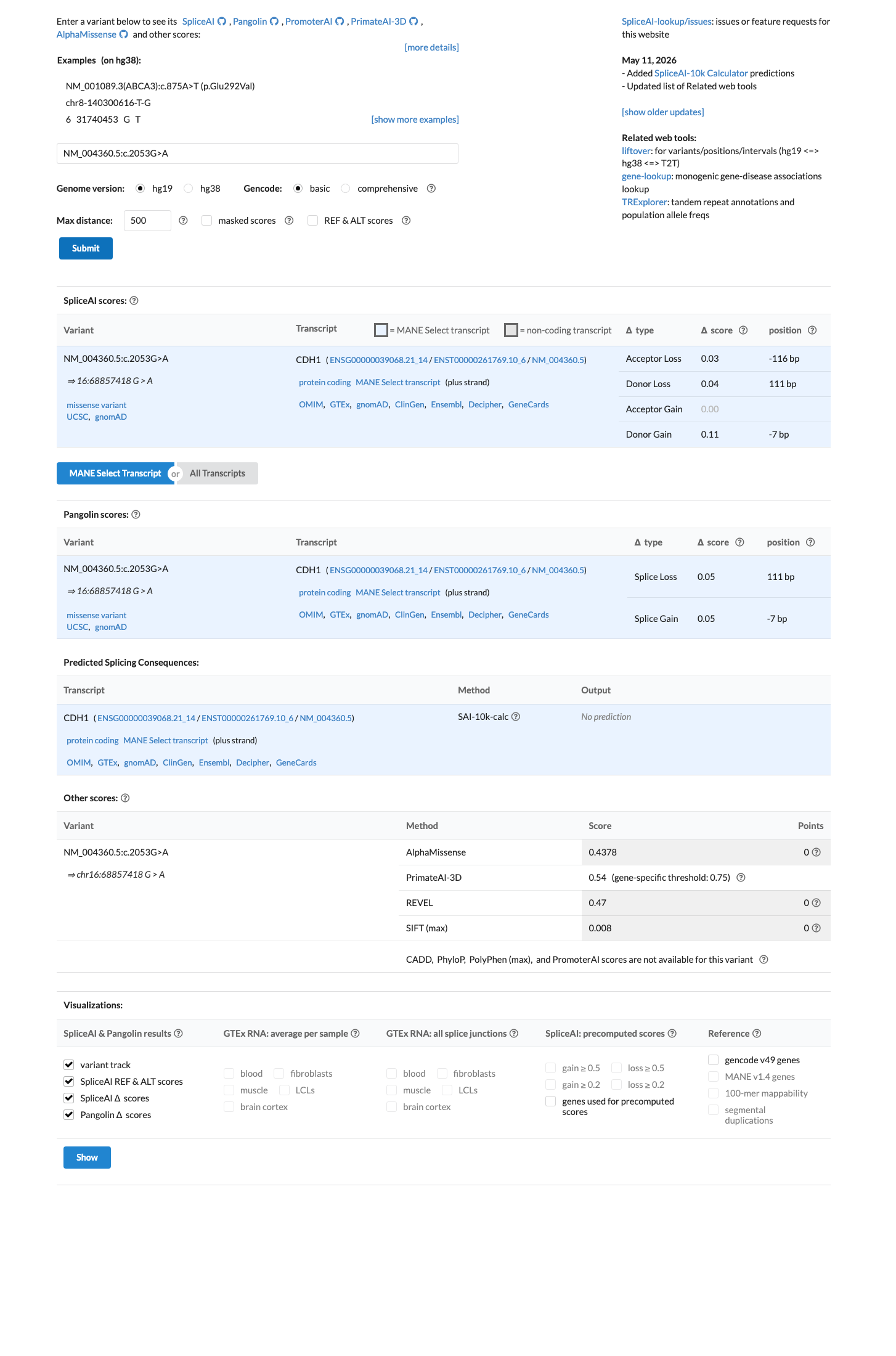
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.11). REVEL score = 0.47. BayesDel score = -0.1394.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. CDH1 (E-cadherin), a tumor suppressor involved in cell adhesion, is altered by mutation or deletion in various cancer typess, most frequently in breas
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
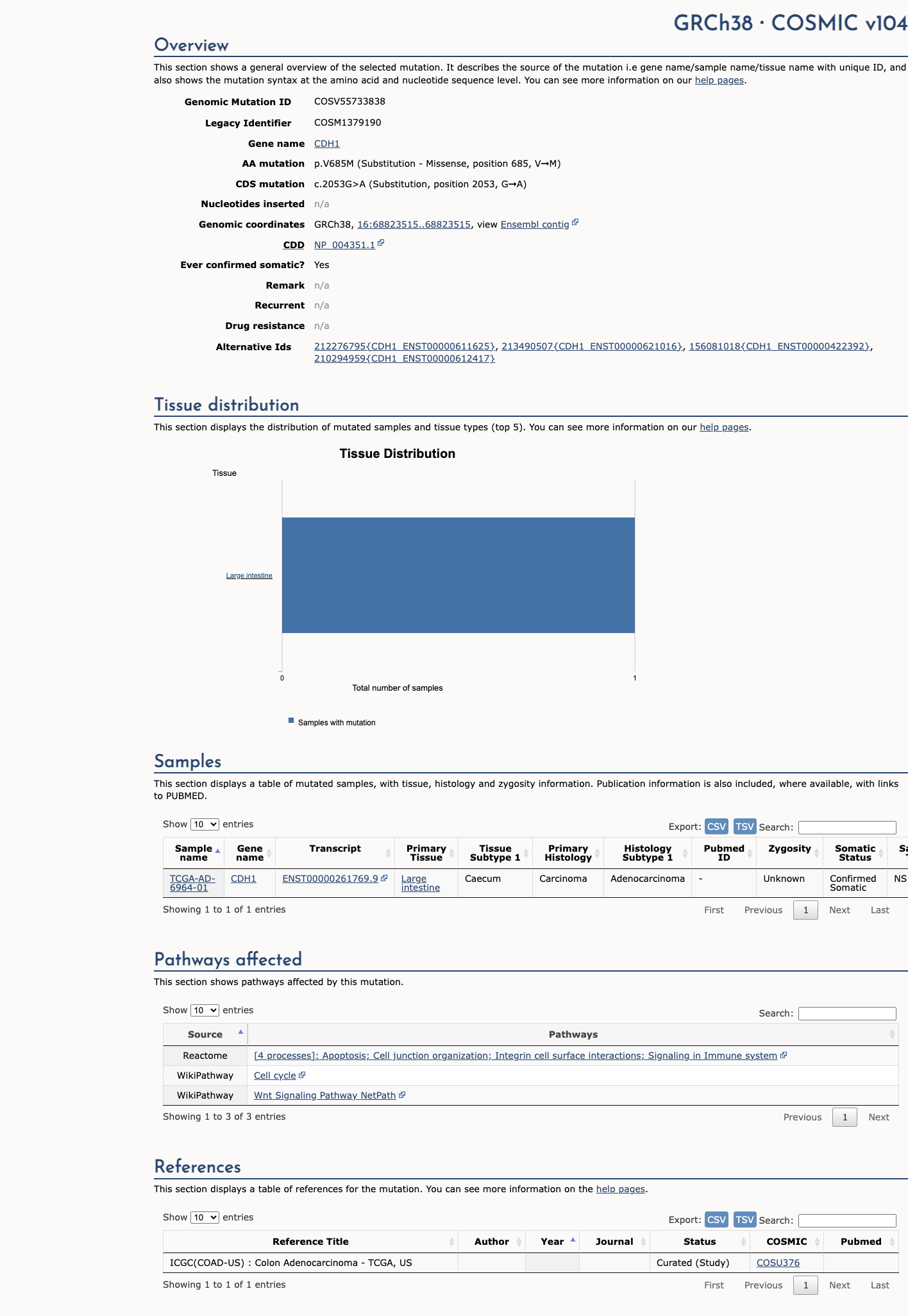
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV55733838, n = 1 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
9Sources
Triaged references · 9 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
29936259 ↗
Analytical Validation of a Hybrid Capture-Based Next-Generation Sequencing Clinical Assay for Genomic Profiling of Cell-Free Circulating Tumor DNA.
CLINVAR
31925297 ↗
Diversity spectrum analysis identifies mutation-specific effects of cancer driver genes.
CLINVAR
17392385 ↗
American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography.
CLINVAR
20065170 ↗
American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility.
CLINVAR
24728327 ↗
Germline variation in cancer-susceptibility genes in a healthy, ancestrally diverse cohort: implications for individual genome sequencing.
CLINVAR
26324357 ↗
American Society of Clinical Oncology Policy Statement Update: Genetic and Genomic Testing for Cancer Susceptibility.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR