NM_004407.4:c.273del (p.Ser92ProfsTer141) is a frameshift deletion in exon 6 of DMP1 predicted to remove over 80% of the protein. DMP1 loss of function is an established mechanism for autosomal recessive hypophosphatemic rickets type 1 (ARHR1), satisfying PVS1 at strong strength under ClinGen SVI PVS1 guidelines (PMC6185798); the variant is in the last exon and NMD is not predicted, warranting a one-level downgrade from very strong.1 This variant is extremely rare in population databases: absent from gnomAD v2.1 and present at 0.00050% in gnomAD v4.1 (8 of 1,614,126 alleles, no homozygotes), meeting PM2 at supporting level.2 SpliceAI predicts no splice-altering impact (max delta = 0.07). REVEL and BayesDel scores are not available for this non-SNV variant. PP3 is not met.3 This variant has been reported in ClinVar as Pathogenic by a single clinical laboratory (Labcorp Genetics/Invitae, SCV007399047). The submitter's cited evidence could not be independently confirmed: PMID:19796717 reports a different DMP1 variant (c.485Tdel) and does not mention c.273del; PMID:28492532 is a methodology paper with no variant-specific content. PP5 is not met.4 Overall classification: Variant of Uncertain Significance (VUS). One strong criterion (PVS1) and one supporting criterion (PM2) are met. Under generic ACMG/AMP 2015 combination rules, this does not reach the threshold for Likely Pathogenic (which requires PVS1_Strong combined with at least 1 moderate or at least 2 supporting criteria). Additional evidence including variant-specific functional studies, case observations, or cosegregation data would be needed to upgrade this classification.5
DMP1
Final classification
VUS
DMP1 c.273del · p.Ser92ProfsTer141
DMP1
NM_004407.4:c.273del (p.Ser92ProfsTer141) is a frameshift deletion in exon 6 of DMP1 predicted to remove over 80% of the protein. DMP1 loss of function is an established mechanism for autosomal recessive hypophosphatemic rickets type 1 (ARHR1), satisfying PVS1 at strong strength under ClinGen SVI PVS1 guidelines (PMC6185798); the variant is in the last exon and NMD is not predicted, warranting a one-level downgrade from very strong.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 strong, PM2 supporting; combination = 1 strong + 1 supporting, which maps to VUS.
Classification rationale
PVS1PM2
VUS
DMP1 c.273del
PVS1 + PM2
→
VUS
1
pvs1_gene_contextpvs1_variant_assessmentpvs1_generic_framework ↗
5
generic_acmg_combination_rules
Gene diagram
· NM_004407.4 · variants mapped to exon structure
DMP1
NM_004407.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 17 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
strong
Pathogenic
NM_004407.4:c.273del is a frameshift variant in exon 6 (last exon) of DMP1, predicted to result in a premature termination codon at p.(Ser92ProfsTer141). DMP1 loss of function is an established mechanism for autosomal recessive hypophosphatemic rickets type 1 (ARHR1), supported by multiple germline disease-focused publications. Under ClinGen SVI PVS1 recommendations (PMC6185798), the variant is in the last exon and is not predicted to undergo nonsense-mediated decay, but removes over 80% of the protein; downgraded from very strong to strong.
Frameshift variant in DMP1 (MANE Select transcript NM_004407.4) resulting in p.Ser92ProfsTer141removing >80% of the 514-amino-acid proteinDMP1 LoF mechanism firmly established for autosomal recessive hypophosphatemic rickets type 1 (ARHR1)
✓
PM2
supporting
Pathogenic
This variant is extremely rare in population databases. It is absent from gnomAD v2.1 and present at very low frequency in gnomAD v4.1 (AF= 4.96e-06, 8/1,614,126 alleles, 0 homozygotes), well below the 0.1% PM2 threshold. Highest subpopulation frequency is European (non-Finnish) at AF= 6.78e-06.
Absent from gnomAD v2.1 (exome)gnomAD v4.1: 8/1614
Assessed · not applied
Pathogenic
PS2
No de novo observation with confirmed parentage has been reported for this variant.
PS3
No well-established functional studies demonstrating a damaging effect specific to NM_004407.4:c.273del have been identified.
PS4
Insufficient data to demonstrate statistically significant enrichment in affected individuals compared to controls.
PM1
This variant does not reside within a known mutational hotspot or critical functional domain as assessed by cancerhotspots.org and domain analysis.
PM6
No assumed de novo observation has been reported for this variant.
PP1
No cosegregation data available for NM_004407.4:c.273del with disease in multiple affected family members.
PP3
No computational evidence supports a deleterious effect for this variant.
PP4
No patient-specific phenotype or family history data are available for assessment.
PP5
This variant has been reported as Pathogenic in ClinVar by a single clinical laboratory (Labcorp Genetics/Invitae, SCV007399047, criteria provided, single submitter).
Benign
BA1
This variant is not present at an allele frequency >1% in any population in gnomAD.
BS1
This variant is not present at an allele frequency >0.3% in any population in gnomAD.
BS2
No healthy adult homozygous individuals have been observed for this variant in population databases (gnomAD homozygote count = 0).
BS3
No well-established in vitro or in vivo functional studies demonstrate no deleterious effect for this variant.
BS4
No evidence of non-segregation with disease has been reported for this variant.
BP2
No evidence of this variant observed in trans with a pathogenic variant for a dominant disorder, or in cis with a pathogenic variant, has been identified.
BP5
No report of this variant in a case with an alternate molecular basis for disease has been identified.
BP6
ClinVar reports this variant as Pathogenic (1 submitter), not as benign.
N/A · 9
PS1 · PM3 · PM4 · PM5 · PP2 · BP1 · BP3 · BP4 · BP7
Research & evidence
Population frequency
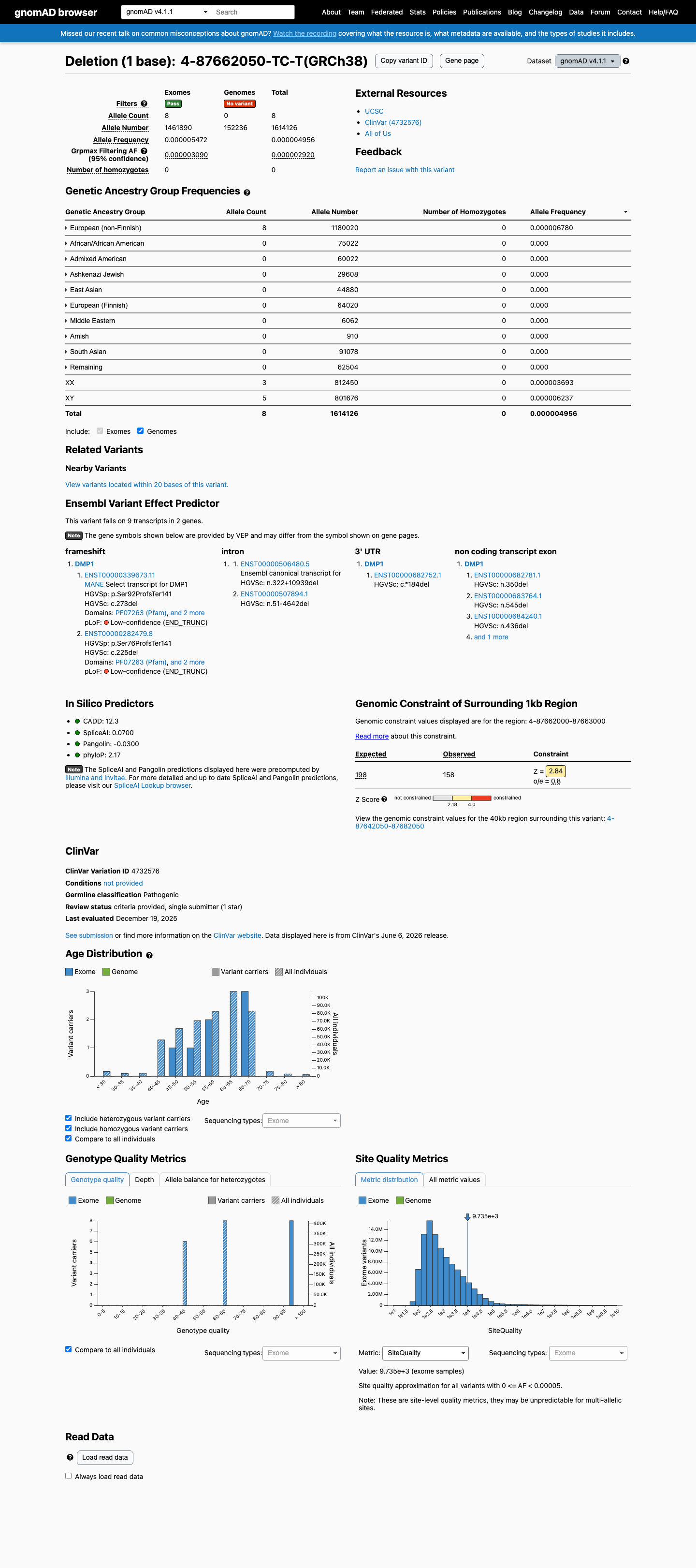
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 4.95624e-06; MAF= 0.00050%, 8/1614126 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 6.77955e-06; MAF= 0.00068%, 8/1180020 alleles, homozygotes = 0); grpmax FAF= 2.92e-06.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0005%
· 8 / 1,614,126
0 hom · FAF 0.00029%
0 hom · FAF 0.00029%
European (non-Finnish) 8 / 1,180,020 |
0.00068% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
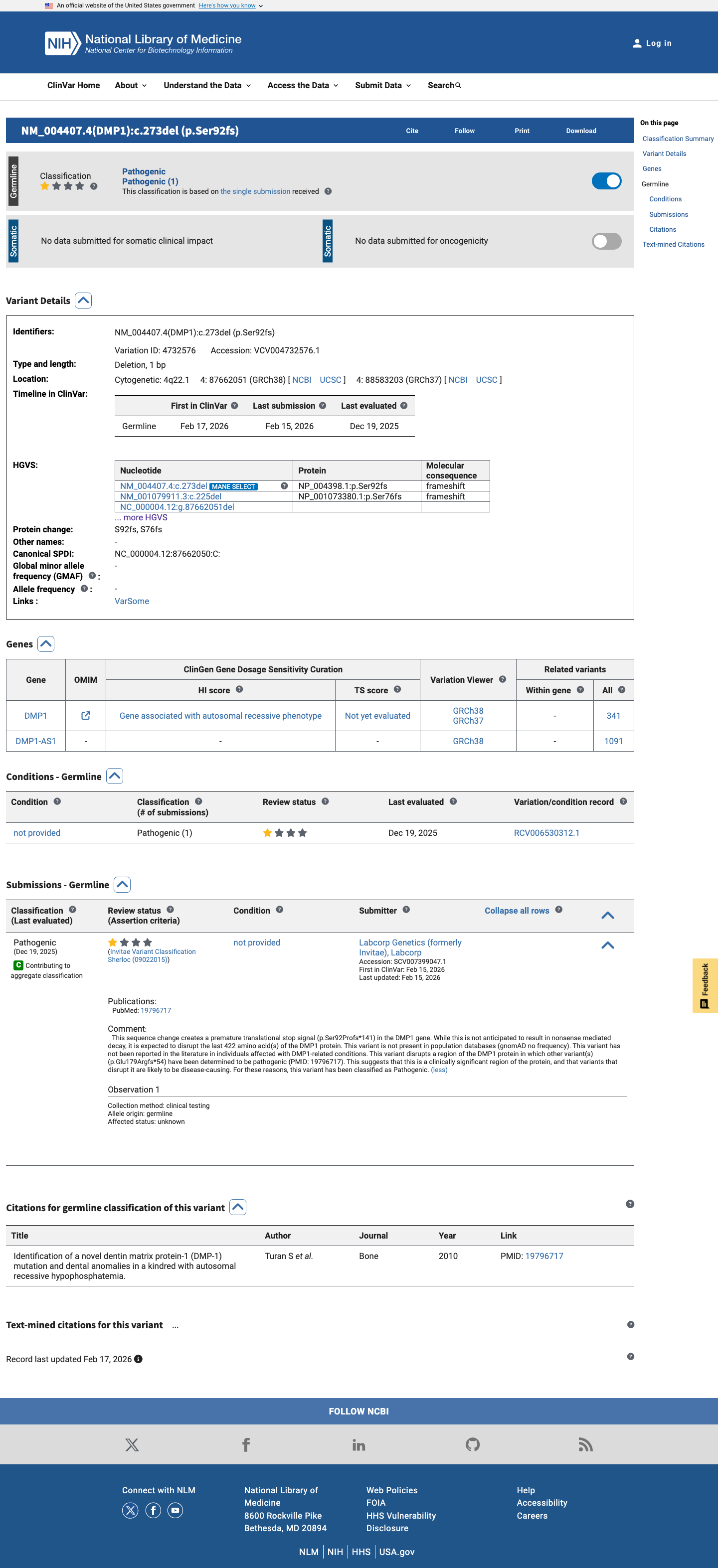
ClinVar
This variant has been reported in ClinVar as Pathogenic (1 clinical laboratory). (ClinVarID = 4732576)
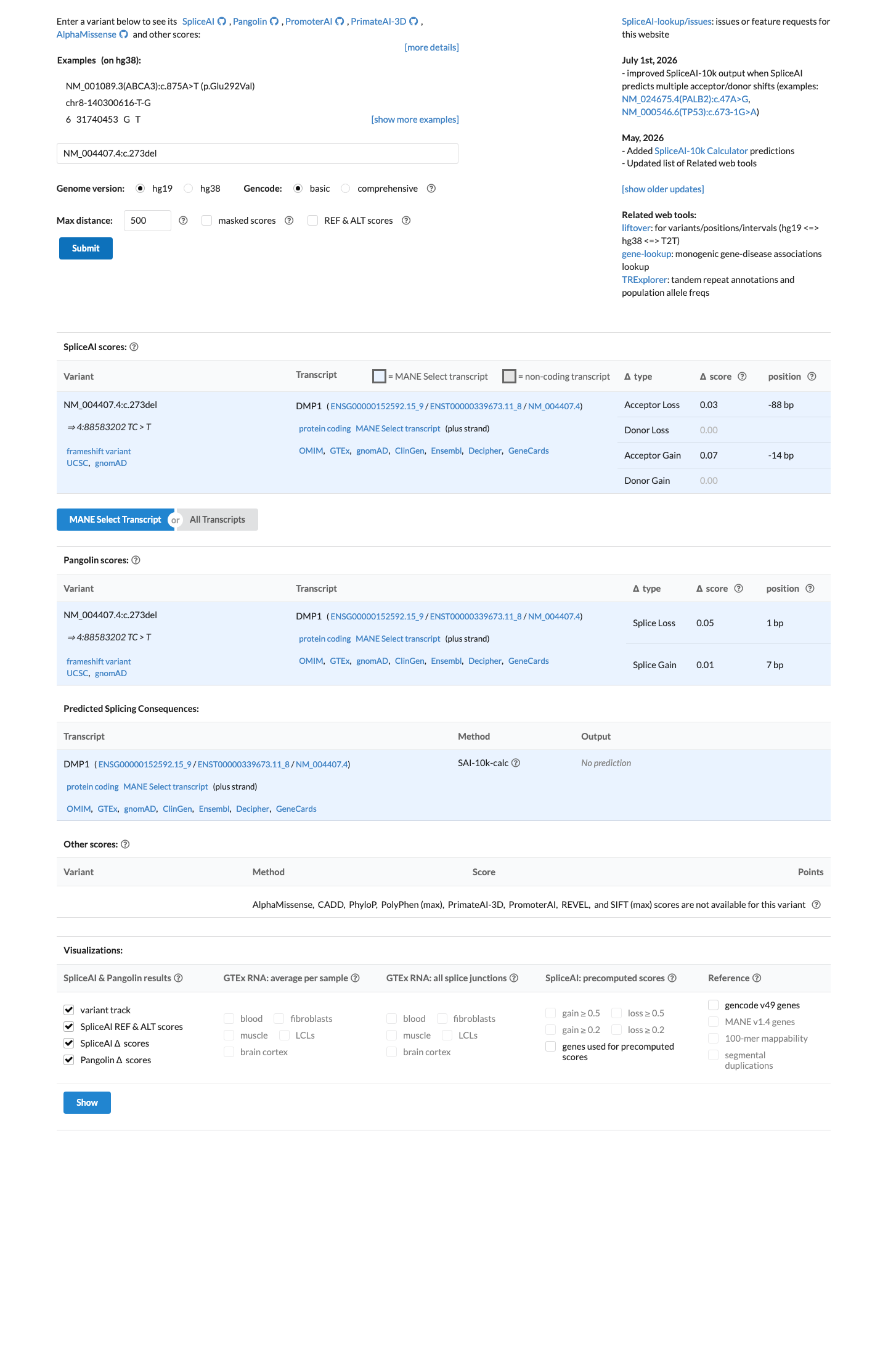
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.07).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 1 PMID not cited in assessment
19796717 ↗
Identification of a novel dentin matrix protein-1 (DMP-1) mutation and dental anomalies in a kindred with autosomal recessive hypophosphatemia.
CLINVAR