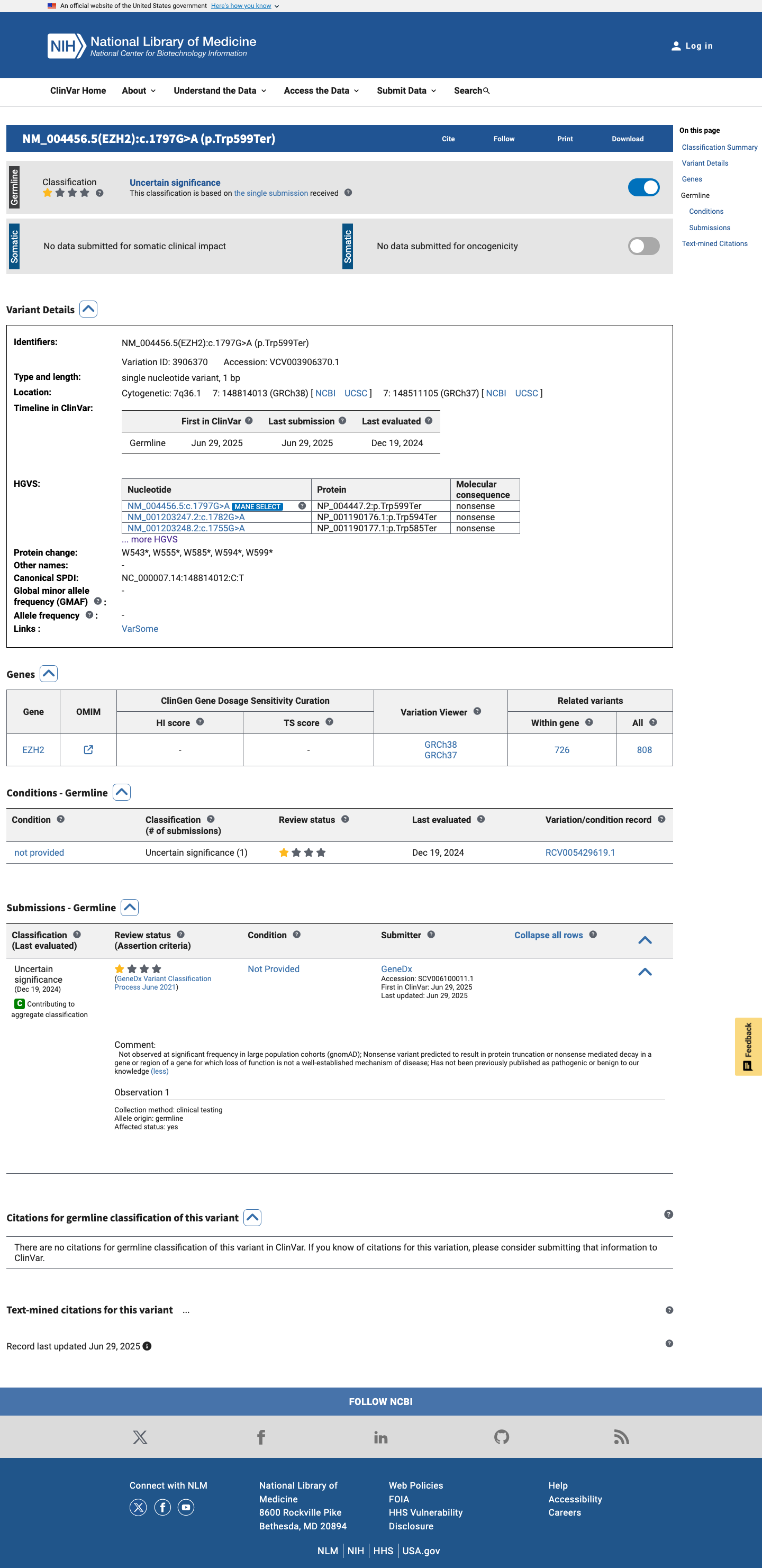
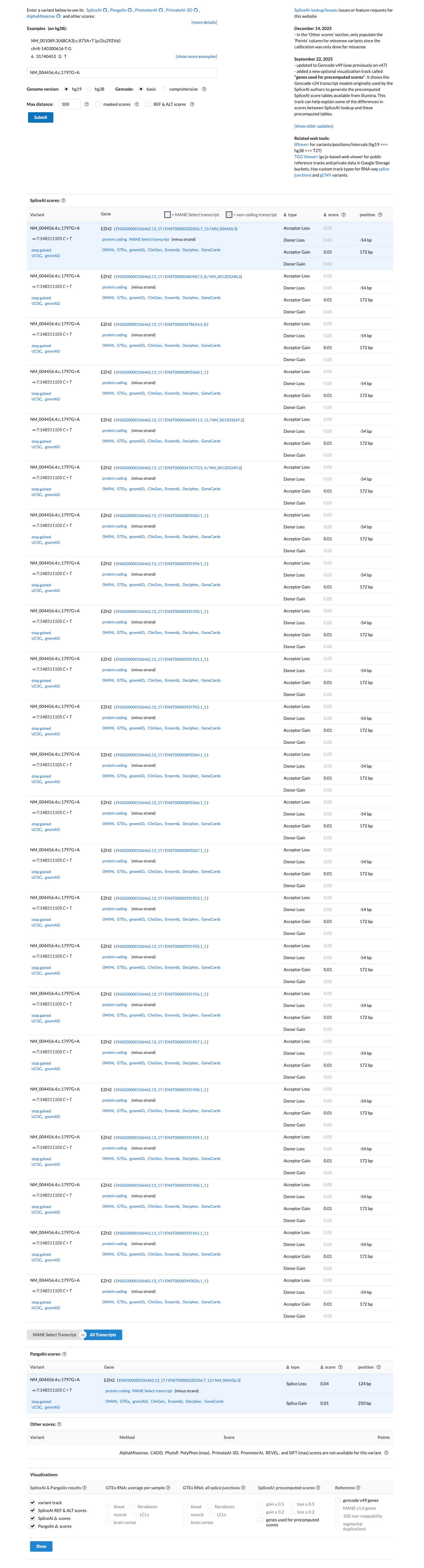
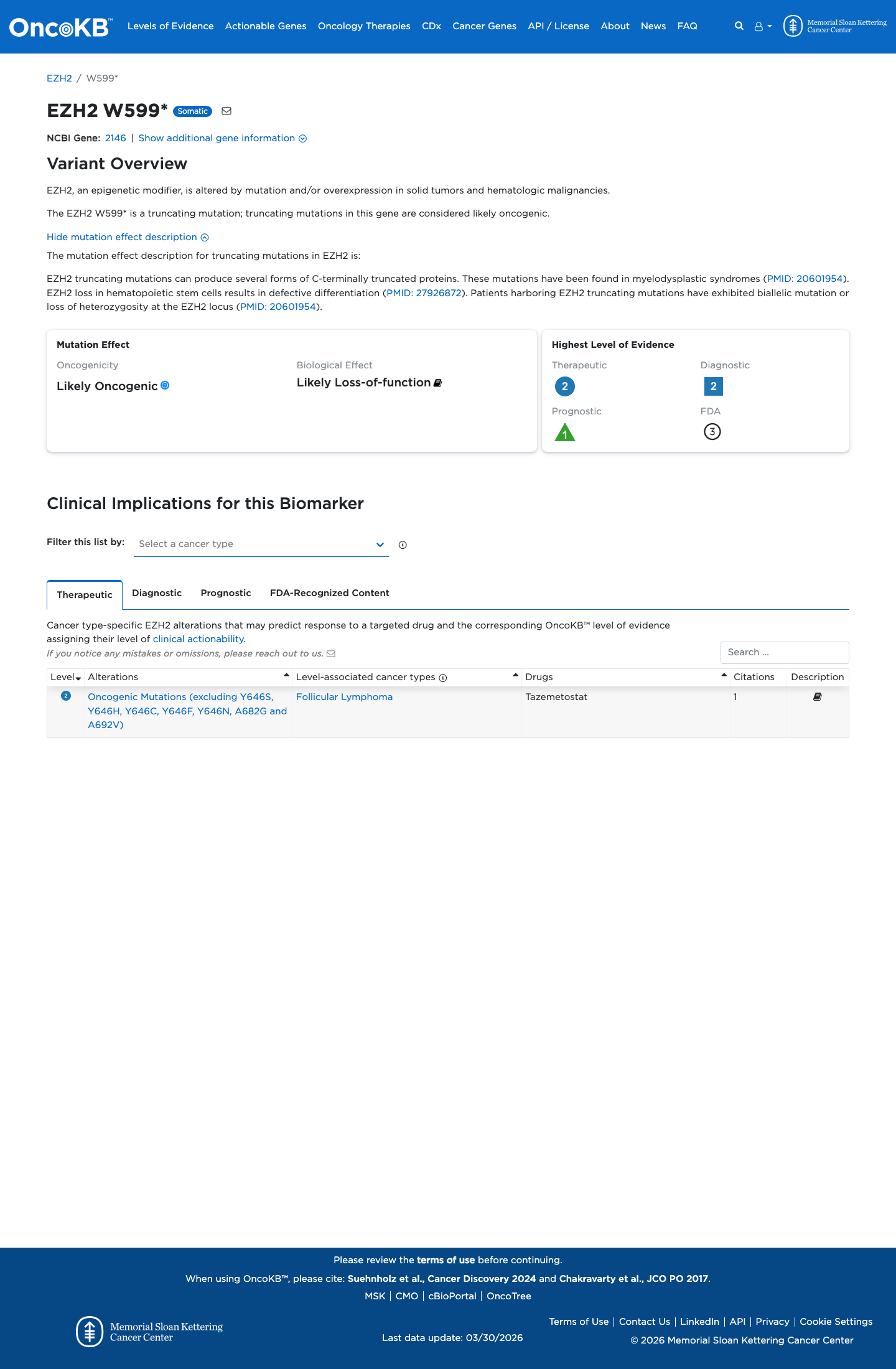
The EZH2 c.1797G>A (p.Trp599Ter; p.W599*) variant has been reported in ClinVar as a variant of uncertain significance with one clinical laboratory submission.1 This variant is absent from gnomAD v2.1 and gnomAD v4.1, which is below the usual PM2 population threshold of 0.1% for a rare germline disorder.2 This nonsense variant is predicted to introduce a premature termination codon in exon 15 with downstream coding exons remaining, and generic PVS1 review indicates that EZH2 is eligible for loss-of-function assessment under the ClinGen SVI framework.3 SpliceAI predicts no significant splice impact for this variant (maximum delta score 0.01), while BayesDel is 0.655873; these computational findings do not provide standalone benign support for this truncating variant.4
EZH2
Final classification
Likely Pathogenic
EZH2 c.1797G>A · p.Trp599Ter
EZH2
The EZH2 c.1797G>A (p.Trp599Ter; p.W599*) variant has been reported in ClinVar as a variant of uncertain significance with one clinical laboratory submission.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 very strong, PM2 supporting; combination = 1 very strong + 1 supporting, which maps to Likely Pathogenic.
Classification rationale
PVS1PM2
Likely Pathogenic
EZH2 c.1797G>A
PVS1 + PM2
→
Likely Pathogenic
3
pvs1_variant_assessmentpvs1_gene_contextpvs1_generic_framework ↗
4
spliceai ↗bayesdel
Gene diagram
· NM_004456.4 · variants mapped to exon structure
EZH2
NM_004456.4
Fetching transcript structure from UCSC…
Applied criteria · 2 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Uncertain significance (1 clinical laboratory).
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). BayesDel score = 0.655873.
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links