Classification rationale
PM2
BP4BP6
Likely Benign
BAP1 c.37+28G>A
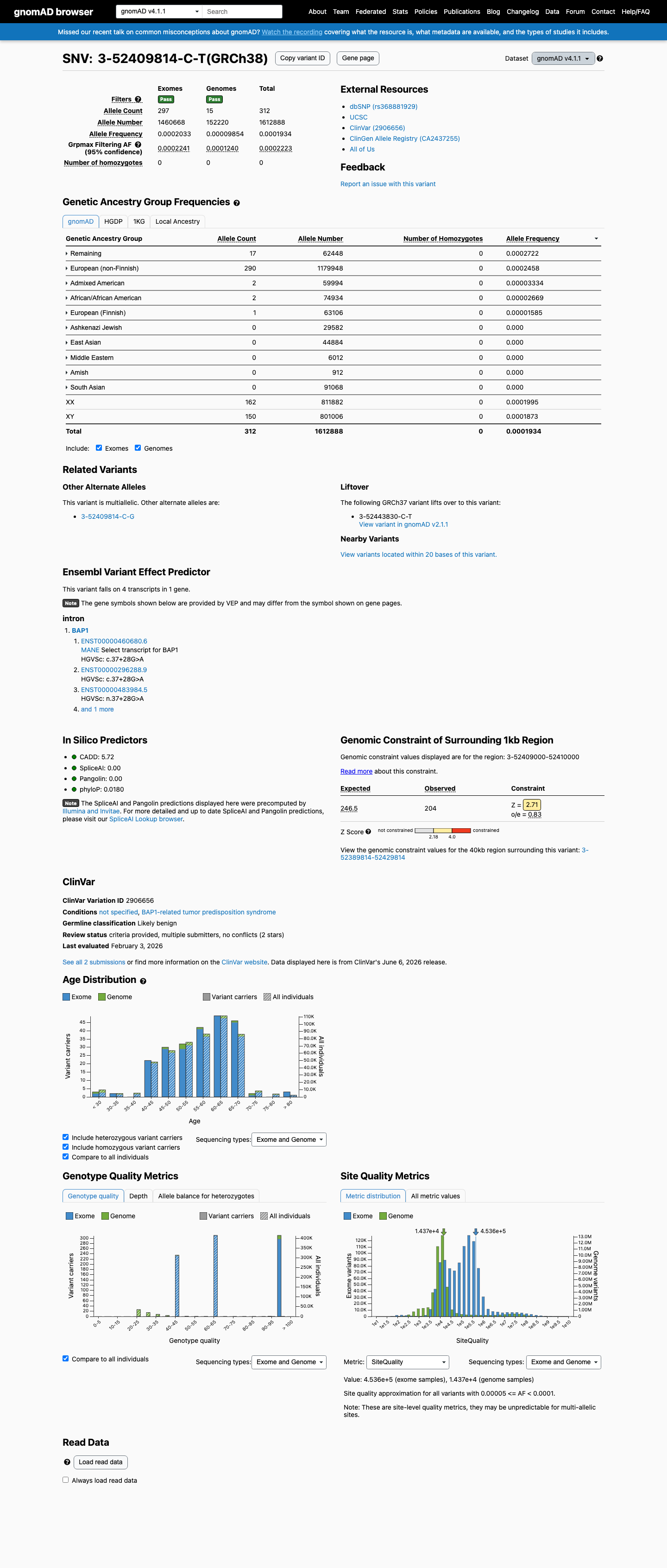
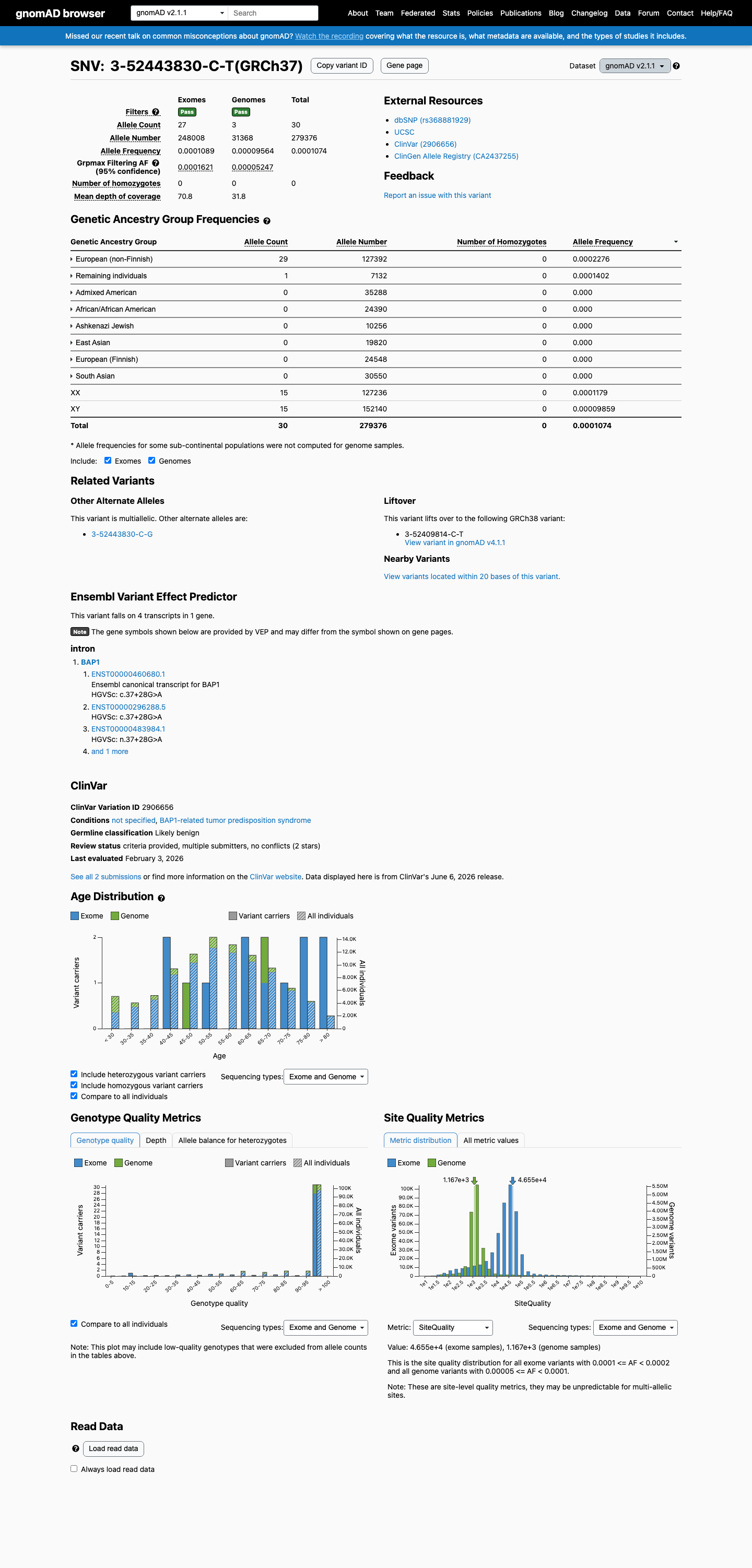
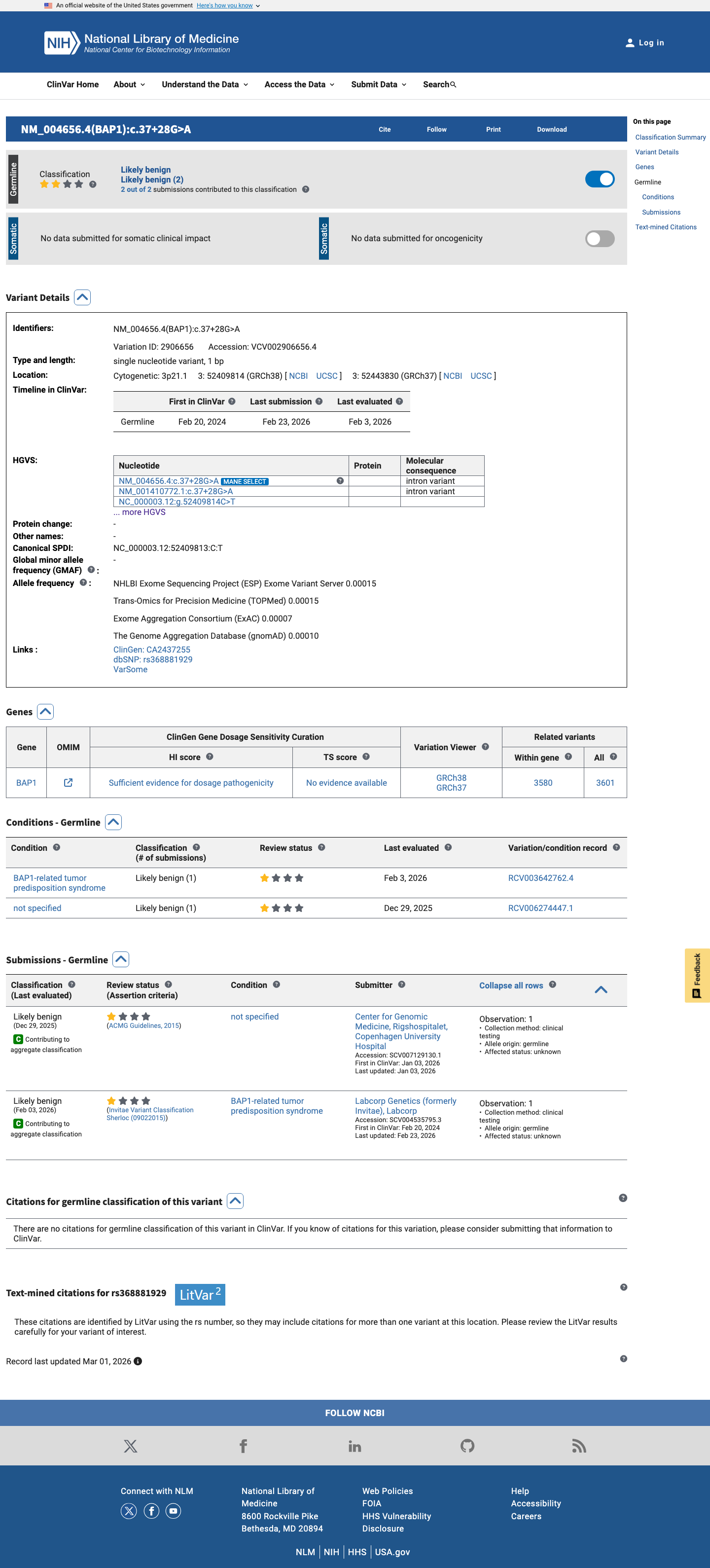
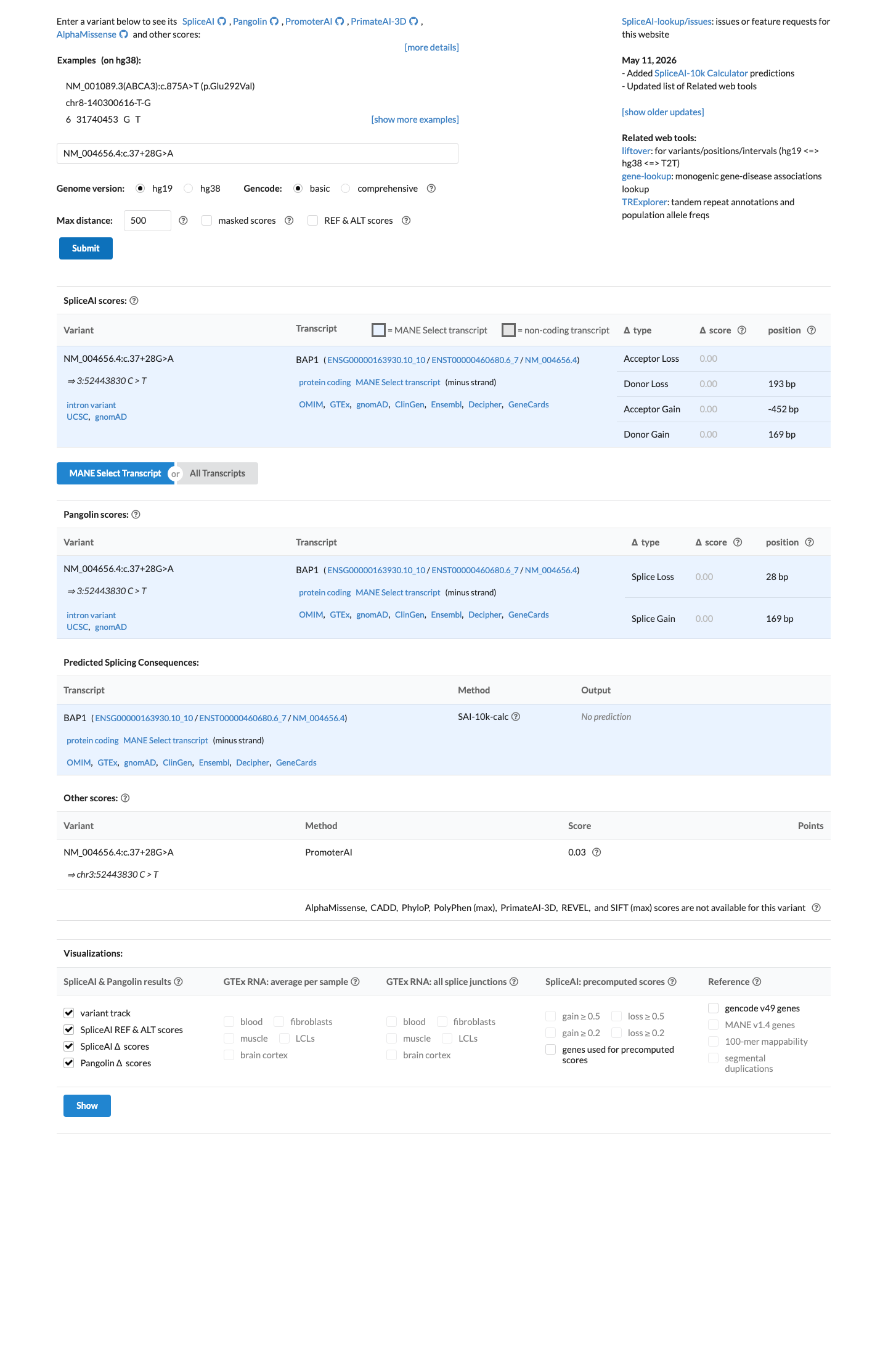
This variant is an intronic substitution (c.37+28G>A) in intron 1 of BAP1. SpliceAI predicts no splice impact (max delta = 0.00).1 Two reputable clinical laboratories have independently classified this variant as Likely benign in ClinVar (Variation ID: 2906656).2 The variant is present in gnomAD at low frequency: 0.0107% in v2.1 (30/279376 alleles) and 0.0193% in v4.1 (312/1612888 alleles), with no homozygotes in any database. This frequency is below the PM2 threshold of 0.1%.3 Applying generic ACMG/AMP 2015 combination rules: one supporting pathogenic criterion (PM2) is outweighed by two supporting benign criteria (BP4, BP6), supporting a Likely benign classification.4
PM2 + BP4 + BP6
→
Likely Benign