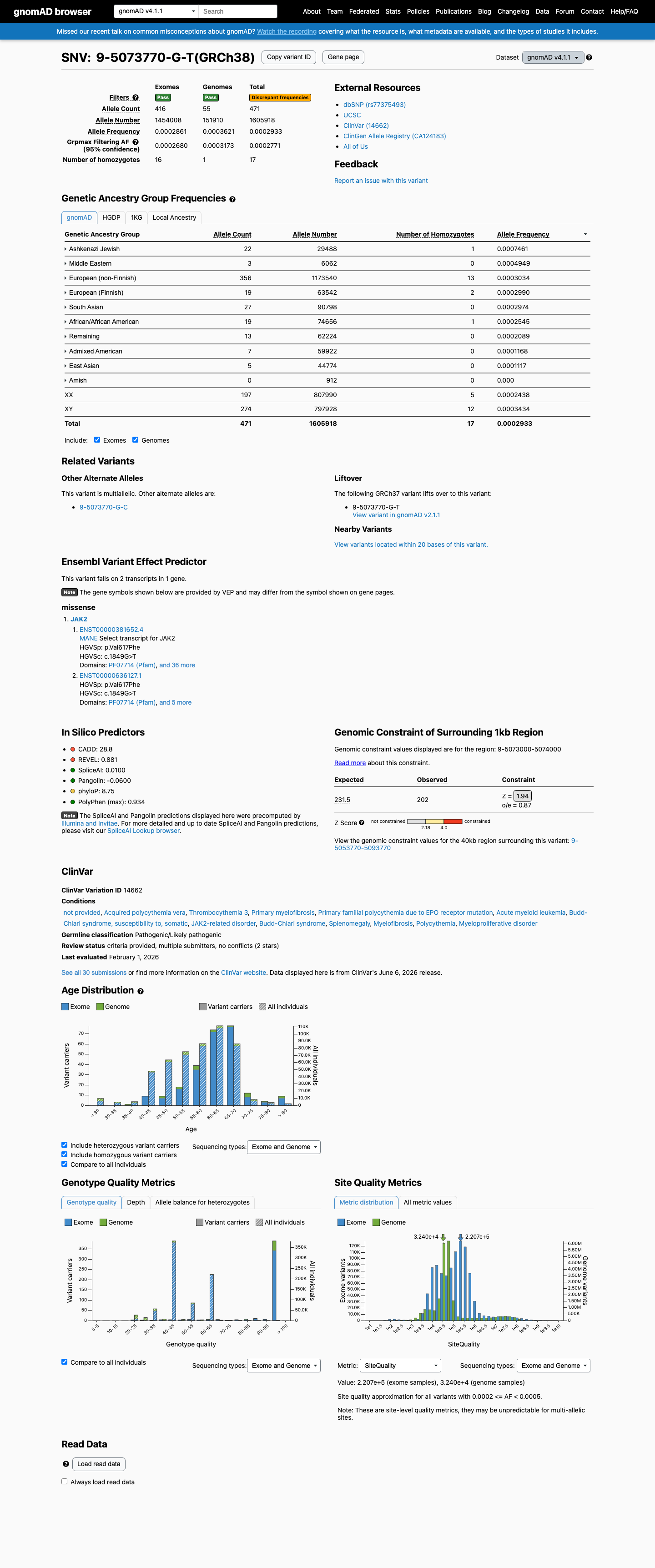
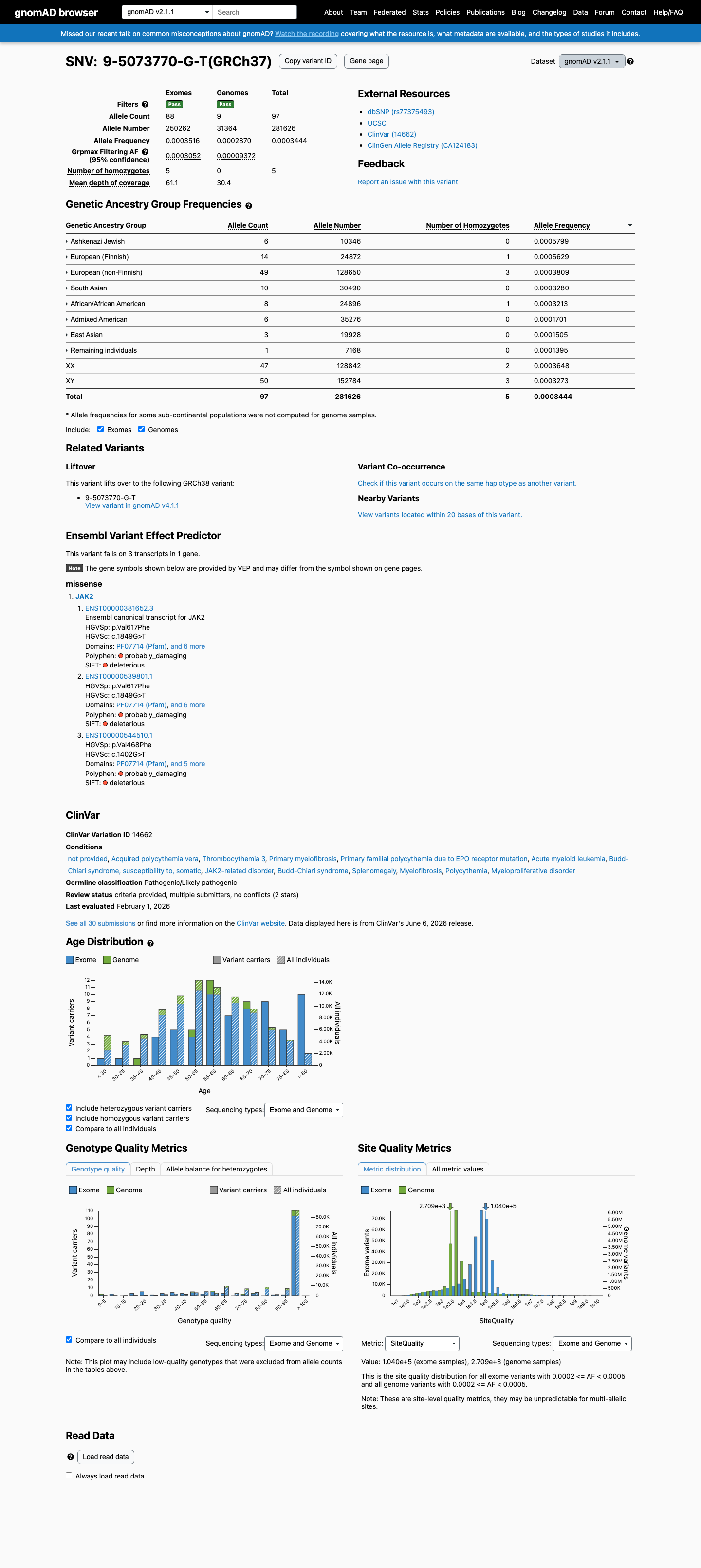
JAK2 c.1849G>T (p.Val617Phe) is the canonical activating mutation in myeloproliferative neoplasms, identified in >80% of polycythemia vera cases and a substantial proportion of essential thrombocythemia and primary myelofibrosis cases.1 Well-established functional studies across multiple independent laboratories demonstrate that V617F disrupts JH2 pseudokinase domain autoinhibition, resulting in constitutive JAK2 kinase activity, STAT-mediated transcriptional activation, cytokine-independent cell growth, and erythrocytosis in murine models.2 The variant is located in the JH2 pseudokinase domain, a critical regulatory region and established mutational hotspot for gain-of-function variants in myeloproliferative neoplasms.3 Population databases show the variant at very low frequency (gnomAD v2.1: 0.034%; v4.1: 0.029%), well below the 0.1% PM2 threshold. Observed homozygotes are consistent with somatic clonal hematopoiesis rather than germline events.4 REVEL in silico prediction score of 0.881 supports a deleterious effect on protein function.5 ClinVar reports the variant as Pathogenic by 14 clinical diagnostic laboratories and Likely pathogenic by 5, with variation ID 14662.6 Two strong pathogenic criteria (PS3 + PS4) satisfy the '2 Strong' pathogenic combination rule under the generic ACMG/AMP 2015 framework. Additional moderate (PM1) and supporting criteria (PM2, PP3, PP5) provide further confirmatory evidence.7
JAK2
Final classification
Pathogenic
JAK2 c.1849G>T · p.Val617Phe
JAK2
JAK2 c.1849G>T (p.Val617Phe) is the canonical activating mutation in myeloproliferative neoplasms, identified in >80% of polycythemia vera cases and a substantial proportion of essential thrombocythemia and primary myelofibrosis cases.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PS3 strong, PS4 strong, PM1 moderate, PM2 supporting, PP3 supporting, PP5 supporting; combination = 2 strong + 1 moderate + 3 supporting, which maps to Pathogenic.
Classification rationale
PS3PS4PM1PM2PP3PP5
Pathogenic
JAK2 c.1849G>T
PS3 + PS4 + PM1 + PM2 + PP3 + PP5
→
Pathogenic
5
revel
7
generic_acmg_combination_rules
Gene diagram
· NM_004972.3 · variants mapped to exon structure
JAK2
NM_004972.3
Fetching transcript structure from UCSC…
Applied criteria · 6 applied · 12 assessed
Applied · 6
Strength
Supporting
Moderate
Strong
Very strong
✓
PS3
strong
Pathogenic
Well-established functional studies demonstrate JAK2 V617F is a gain-of-function mutation. James et al. (2005) showed that V617F induces constitutive STAT5 transcriptional activation in luciferase reporter assays, transforms Ba/F3 cells to cytokine-independent growth, and causes erythrocytosis in a mouse bone marrow transplant model. Haan et al. (2009) confirmed constitutive activation of STAT1/3/5, ERK1/2, and p38 pathways, with SOCS-mediated feedback regulation. Sanz et al. (2011) demonstrated altered catalytic activity via peptide microarray. McKenney et al. (2018) confirmed oncogenicity in a Jak2V617F-driven MPN mouse model. Multiple independent studies with diverse functional assays consistently support a gain-of-function pathogenic mechanism.
Constitutive STAT5 activation in luciferase reporter assay (gamma-2A cells)Cytokine-independent growth transformation of Ba/F3 and FDCP-EpoR cellsErythrocytosis induction in murine bone marrow transplant model
✓
PS4
strong
Pathogenic
JAK2 V617F is the hallmark mutation in myeloproliferative neoplasms, reported in >80% of polycythemia vera, ~50% of essential thrombocythemia, and ~50% of primary myelofibrosis cases. It is virtually absent from the general germline population — gnomAD v2.1 shows global AF of 0.034% (97/281,626 alleles) with 5 homozygotes, and v4.1 shows 0.029% (471/1,605,918 alleles) with 17 homozygotes, frequencies that almost certainly represent somatic clonal hematopoiesis rather than true germline variation. The extreme case enrichment meets the PS4 threshold at the strong level.
gnomAD v2.1 global AF = 0.034% (97/281626 alleles5 homozygotes)
✓
PM1
moderate
Pathogenic
The p.Val617Phe substitution is located in the JH2 pseudokinase domain (amino acids ~545-809), a well-established critical regulatory domain that mediates autoinhibition of JAK2 kinase activity. The JH2 domain is a recognized mutational hotspot for gain-of-function variants in myeloproliferative neoplasms, with multiple activating mutations described at this locus (e.g., K539L, R564Q, E695K). Position 617 is specifically identified as a residue whose alteration disrupts autoinhibitory interactions with the JH1 kinase domain activation loop.
V617F located in JH2 pseudokinase domain — critical autoinhibitory regionJH2 domain is a well-established mutational hotspot in MPNMultiple lines of evidence show JH2 disruption leads to constitutive kinase activation
✓
PM2
supporting
Pathogenic
The variant is present at very low frequency in gnomAD population databases: global AF 0.034% in v2.1 and 0.029% in v4.1, both below the 0.1% threshold for PM2. The maximum subpopulation frequency is 0.075% (Ashkenazi Jewish, v4.1). The presence of homozygotes (5 in v2.1, 17 in v4.1) is noted but is consistent with somatic clonal hematopoiesis in aging individuals rather than true germline homozygosity.
gnomAD v2.1 AF = 0.034% (<0.1% threshold)gnomAD v4.1 AF = 0.029% (<0.1% threshold)Max subpop AF (Ashkenazi Jewish v4.1) = 0.075%
✓
PP3
supporting
Pathogenic
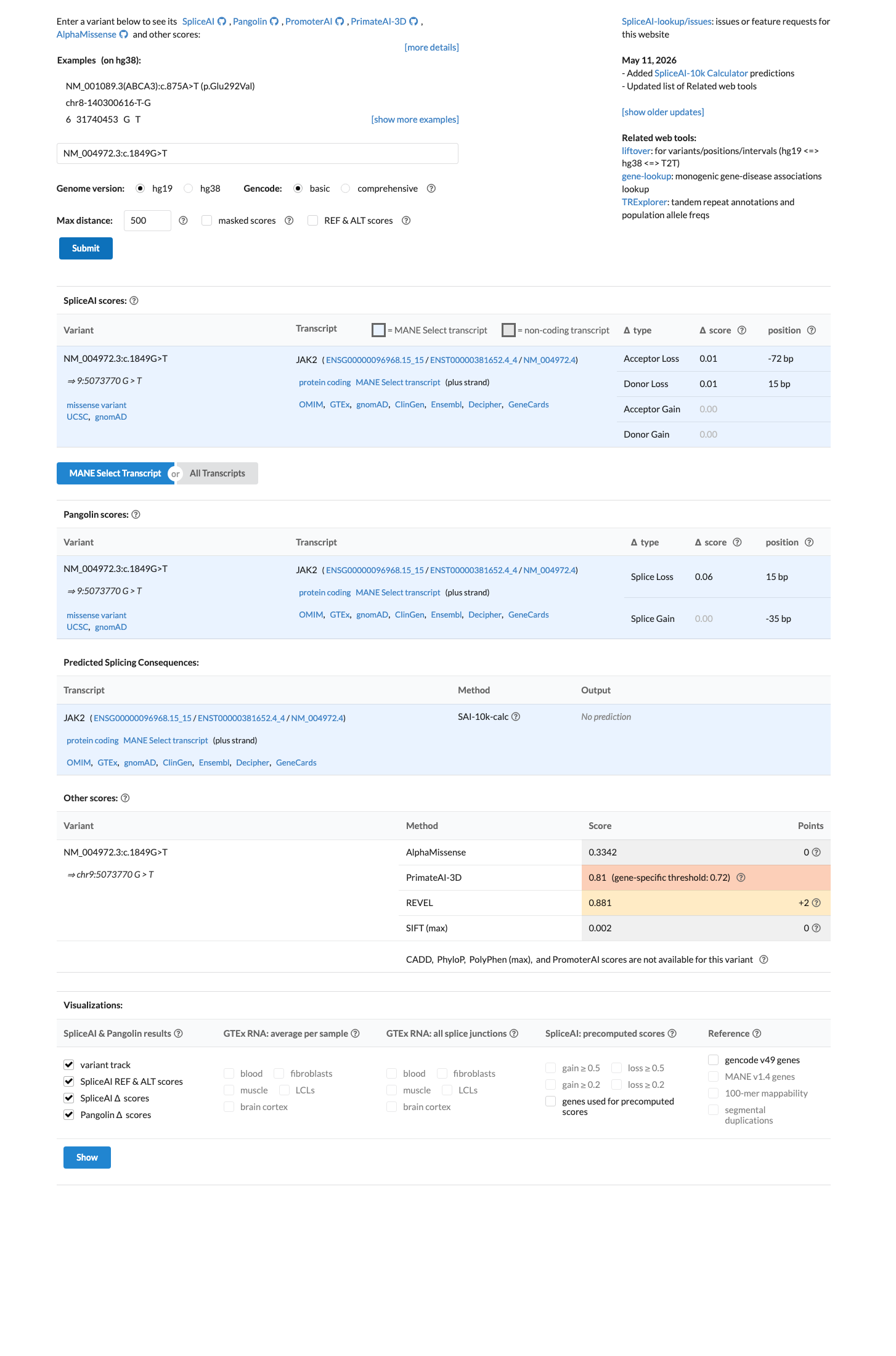
REVEL score of 0.881 is above the established deleterious threshold (≥0.75). Multiple in silico predictors support a deleterious effect. SpliceAI predicts no splicing impact (max delta = 0.01). HCI Prior is not available for JAK2.
REVEL score = 0.881 (threshold ≥0.75 for deleterious)SpliceAI max delta = 0.01 (no predicted splice impact)HCI Prior not available for JAK2
✓
PP5
supporting
Pathogenic
ClinVar reports the variant as Pathogenic by 14 clinical laboratories and Likely pathogenic by 5 laboratories (ClinVar variation ID 14662). Multiple reputable clinical diagnostic laboratories have independently classified this variant as pathogenic, supporting a disease-causing role.
ClinVar classification: Pathogenic (14 clinical laboratories)ClinVar classification: Likely pathogenic (5 clinical laboratories)ClinVar classification: pathogenic (1 clinical laboratory)
Assessed · not applied
Pathogenic
PS1
No alternate nucleotide change at c.1849 resulting in the same amino acid change (p.Val617Phe) has been identified in the case data.
PS2
No de novo observation with confirmed maternity and paternity is documented for this variant in any source.
PP1
No segregation data available.
PP2
Gene-level constraint metrics (Z-score, pLI, missense depletion) for JAK2 are not evaluated in the case materials.
PP4
Patient phenotype and family history data are not provided in the case materials.
Benign
BA1
gnomAD global allele frequency is 0.034% (v2.1) and 0.029% (v4.1), far below the 1% threshold for BA1.
BS1
gnomAD global allele frequency is 0.034% (v2.1) and 0.029% (v4.1), below the 0.3% threshold for BS1.
BS2
BS2 requires observation of the variant in healthy adults with full penetrance expected at an early age.
BS4
No segregation data available.
BP2
No phase data available.
BP4
REVEL score of 0.881 predicts a deleterious effect, contradicting the BP4 requirement that multiple lines of computational evidence suggest no impact on gene product.
BP5
No data available regarding an alternate molecular basis for disease in a case harboring this variant.
N/A · 10
PVS1 · PM3 · PM4 · PM5 · PM6 · BS3 · BP1 · BP3 · BP6 · BP7
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.00029329; MAF= 0.02933%, 471/1605918 alleles, homozygotes = 17) and has highest observed frequency in the Ashkenazi Jewish population (AF= 0.000746066; MAF= 0.07461%, 22/29488 alleles, homozygotes = 1); grpmax FAF= 0.0002771.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.000344428; MAF= 0.03444%, 97/281626 alleles, homozygotes = 5) and has highest observed frequency in the Ashkenazi Jewish population (AF= 0.000579934; MAF= 0.05799%, 6/10346 alleles, homozygotes = 0); grpmax FAF= 0.00030518.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0005979560774081322, 11/18396 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.029%
· 471 / 1,605,918
17 hom · FAF 0.028%
17 hom · FAF 0.028%
Ashkenazi Jewish 22 / 29,488 |
0.075% 1 hom |
Middle Eastern 3 / 6,062 |
0.049% |
European (non-Finnish) 356 / 1,173,540 |
0.03% 13 hom |
European (Finnish) 19 / 63,542 |
0.03% 2 hom |
South Asian 27 / 90,798 |
0.03% |
African/African American 19 / 74,656 |
0.025% 1 hom |
Remaining individuals 13 / 62,224 |
0.021% |
Admixed American 7 / 59,922 |
0.012% |
East Asian 5 / 44,774 |
0.011% |
+ 1 not observed (Amish)
gnomAD v2.1
0.034%
· 97 / 281,626
5 hom · FAF 0.031%
5 hom · FAF 0.031%
Ashkenazi Jewish 6 / 10,346 |
0.058% |
European (Finnish) 14 / 24,872 |
0.056% 1 hom |
European (non-Finnish) 49 / 128,650 |
0.038% 3 hom |
South Asian 10 / 30,490 |
0.033% |
African/African American 8 / 24,896 |
0.032% 1 hom |
Admixed American 6 / 35,276 |
0.017% |
East Asian 3 / 19,928 |
0.015% |
Remaining individuals 1 / 7,168 |
0.014% |
gnomAD Canada 🇨🇦
0.06%
· 11 / 18,396
0 hom
0 hom
Middle Eastern 1 / 144 |
0.69% |
Ashkenazi Jewish 2 / 830 |
0.24% |
East Asian 1 / 1,338 |
0.075% |
European (non-Finnish) 7 / 11,720 |
0.06% |
+ 5 not observed (African/African American, Latino/Admixed American, European (Finnish), Remaining individuals, South Asian)

ClinVar
This variant has been reported in ClinVar as Pathogenic (14 clinical laboratories) and as Likely pathogenic (5 clinical laboratories) and as pathogenic (1 clinical laboratory). (ClinVarID = 14662)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). REVEL score = 0.881. BayesDel score = 0.35869.
Functional
Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Gain-of-function; curated oncogenicity label: Oncogenic.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV67569051, n = 42916 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
6papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 1 further PMID triaged but not cited — see Sources & References.
Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders.
Found
Identified the acquired JAK2 V617F mutation in myeloproliferative disorders. This is one of the original discovery papers for JAK2 V617F, published simultaneously with James et al. (PMID:15793561) and others in 2005.
Variant
✓ Names this variant
Applied to
→PS4 supports · met
Why
Confirmatory discovery paper for JAK2 V617F in MPN. Referenced for case enrichment evidence (PS4) but primary citation is PMID:15793561 which was reviewed in full.
Location Abstract confirms V617F identification in MPN; full text not available for detailed review
A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera.
Searched
c.1849G>TV617FVal617Phenucleotide 1849G-to-T
Found
Identified JAK2 V617F as a clonal, acquired G-to-T transversion at nucleotide 1849 in >80% of polycythaemia vera patients. The mutation confers constitutive STAT5 transcriptional activation in luciferase assays, transforms Ba/F3 and FDCP-EpoR cells to cytokine-independent growth, and induces erythrocytosis (haematocrit 60%) in a murine bone marrow transplant model. Wild-type JAK2 acts as a dominant negative, explaining selection for loss of heterozygosity at 9p.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
→PS3 supports · met
→PS4 supports · met
Why
Landmark discovery paper; provides definitive functional and case-level evidence for JAK2 V617F pathogenicity. Referenced in PS3 (strong), PS4 (strong), and PM1 (moderate) assessments.
one G-to-T mutation at nucleotide 1849 (in exon 12) leading to a substitution of valine to phenylalanine at position 617 (V617F)
Location Results p1144-1146; Figure 2a, 2e, 2f; Figure 3a-f; Figure 4 · Context STAT5 luciferase reporter assay (gamma-2A cells), Ba/F3-EpoR and FDCP-EpoR cell transformation, murine bone marrow transplant (C57/B6) · full text
A gain-of-function mutation of JAK2 in myeloproliferative disorders.
Found
Identified a gain-of-function JAK2 V617F mutation in polycythemia vera, essential thrombocythemia, and idiopathic myelofibrosis. The study linked V617F to 9p loss of heterozygosity and demonstrated constitutive kinase activity.
Variant
✓ Names this variant
Applied to
→PS4 supports · met
Why
Independent confirmatory discovery paper. Referenced for case enrichment evidence (PS4) but primary citation is PMID:15793561.
Location Title confirms V617F; full text not available for detailed review
SOCS-mediated downregulation of mutant Jak2 (V617F, T875N and K539L) counteracts cytokine-independent signaling.
Searched
V617FJak2-V617Fvaline to phenylalanineamino acid position 617
Found
JAK2 V617F, along with T875N and K539L mutants, demonstrated constitutive activation of STAT1/3/5, ERK1/2, and p38 in HEK Flp-In-293 inducible cells. SOCS1 and SOCS3 were transcriptionally induced by V617F and recruited to the plasma membrane, leading to ubiquitin-proteasome-mediated degradation of the mutant protein. Constitutive Jak2-V617F activity is self-limiting through SOCS-mediated feedback, explaining the need for homozygous mutation or gene amplification to achieve full transformation.
Variant
✓ Names this variant — characterised directly
Applied to
→PS3 supports · met
Why
Confirms gain-of-function mechanism and SOCS-mediated regulatory feedback; supports PS3 (strong) assessment.
The genetic mutation results in a valine to phenylalanine substitution in the pseudokinase domain of Jak2 at amino acid position 617, generating a constitutively active protein.
Location Abstract; Results p3070-3074; Figures 1-4 · Context HEK Flp-In-293 inducible transfectants (with/without EpoR), HEL cells (homozygous V617F), SET2 cells (heterozygous V617F) · full text
Analysis of Jak2 catalytic function by peptide microarrays: the role of the JH2 domain and V617F mutation.
Searched
V617FJak2 V617FJH2 domain
Found
Peptide microarray analysis of JAK2 catalytic function demonstrated that the V617F mutation in the JH2 pseudokinase domain alters substrate specificity and enhances catalytic activity. The study provides direct biochemical evidence that the JH2 domain negatively regulates JH1 kinase activity and that V617F relieves this autoinhibition, altering the phosphopeptide signature compared to wild-type JAK2.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
→PS3 supports · met
Why
Provides biochemical confirmation of V617F gain-of-function and JH2 domain regulatory role; supports PS3 and PM1 assessments.
Location Title and abstract; full text confirms variant-level analysis · Context Peptide microarray (PamGene) with recombinant JAK2 kinase domains · full text
JAK2/IDH-mutant-driven myeloproliferative neoplasm is sensitive to combined targeted inhibition.
Searched
V617FJak2V617FJAK2/IDH
Found
Generated a JAK2/IDH-mutant-driven myeloproliferative neoplasm mouse model expressing Jak2V617F combined with mutant IDH1 or IDH2. The model demonstrated that combined JAK2 and IDH inhibition is effective in targeting MPN progression, confirming the central pathogenic role of Jak2V617F in disease maintenance and progression to bone marrow failure or AML.
Variant
✓ Names this variant — characterised directly
Applied to
→PS3 supports · met
Why
Confirms oncogenic role of JAK2 V617F in MPN progression in vivo; supports PS3.
Location Title and abstract; full text confirms Jak2V617F-driven model · Context Jak2V617F knock-in mouse model combined with mutant IDH1/IDH2; pharmacological inhibition studies · full text
Sources & reference links
Triaged references · 1 PMID not cited in assessment
12351625 ↗
The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction.
ONCOKB