De novo occurrence confirmed in a proband with juvenile polyposis and hereditary hemorrhagic telangiectasia overlap syndrome; variant absent from both unaffected parents with paternity and maternity confirmed by haplotype analysis.1 Observed in at least 2-3 unrelated probands with juvenile polyposis syndrome, representing a significantly increased prevalence in affected individuals compared to the general population.2 Located within the MH2 domain (Mutational Rich Region 1), a well-established functional domain and statistically significant mutational hotspot in SMAD4.3 Absent from gnomAD v2.1, v4.1, and gnomAD-Canada population databases, with allele frequency of zero in all populations.4 REVEL score of 0.885 strongly predicts a deleterious effect; SpliceAI confirms no cryptic splice alteration (max delta 0.05), consistent with a missense mechanism.5 Proband phenotype (colonic juvenile polyps, anemia, telangiectases, epistaxis) is highly specific for the SMAD4-associated JP-HHT overlap syndrome.6
SMAD4
Final classification
Pathogenic
SMAD4 c.1081C>G · p.Arg361Gly
SMAD4
De novo occurrence confirmed in a proband with juvenile polyposis and hereditary hemorrhagic telangiectasia overlap syndrome; variant absent from both unaffected parents with paternity and maternity confirmed by haplotype analysis.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PS2 strong, PS4 moderate, PM1 moderate, PM2 moderate, PP3 supporting, PP4 supporting; combination = 1 strong + 3 moderate + 2 supporting, which maps to Pathogenic.
Classification rationale
PS2PS4PM1PM2PP3PP4
Pathogenic
SMAD4 c.1081C>G
PS2 + PS4 + PM1 + PM2 + PP3 + PP4
→
Pathogenic
Gene diagram
· NM_005359.5 · variants mapped to exon structure
SMAD4
NM_005359.5
Fetching transcript structure from UCSC…
Applied criteria · 6 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
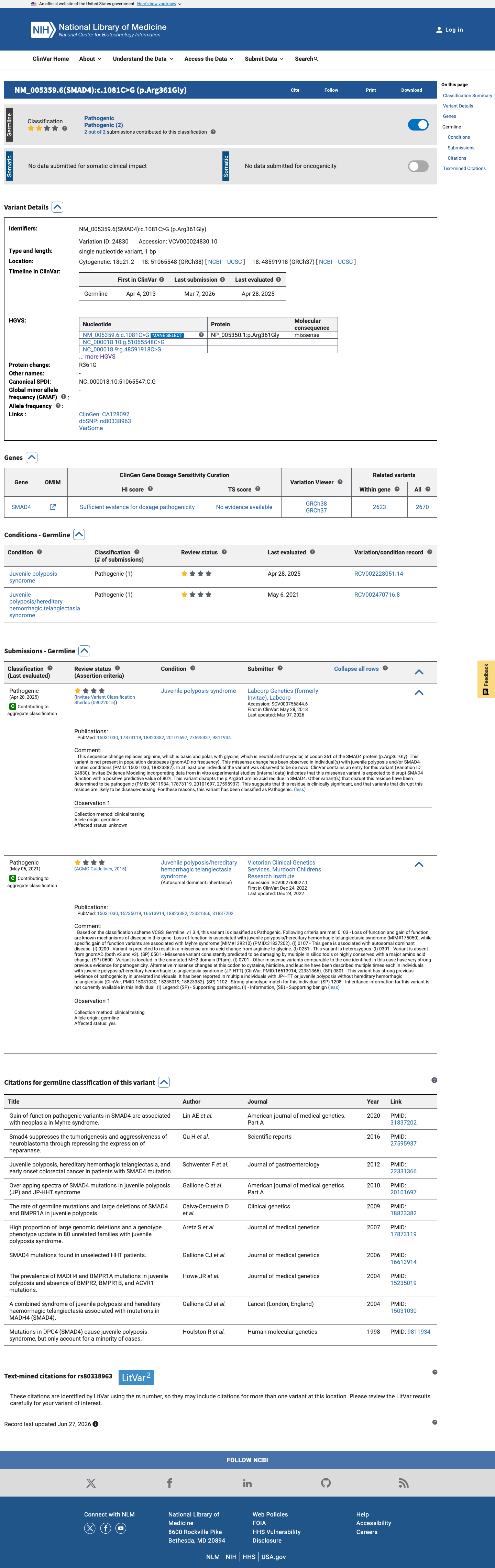
This variant has been reported in ClinVar as Pathogenic (2 clinical laboratories). (ClinVarID = 24830)
In silico
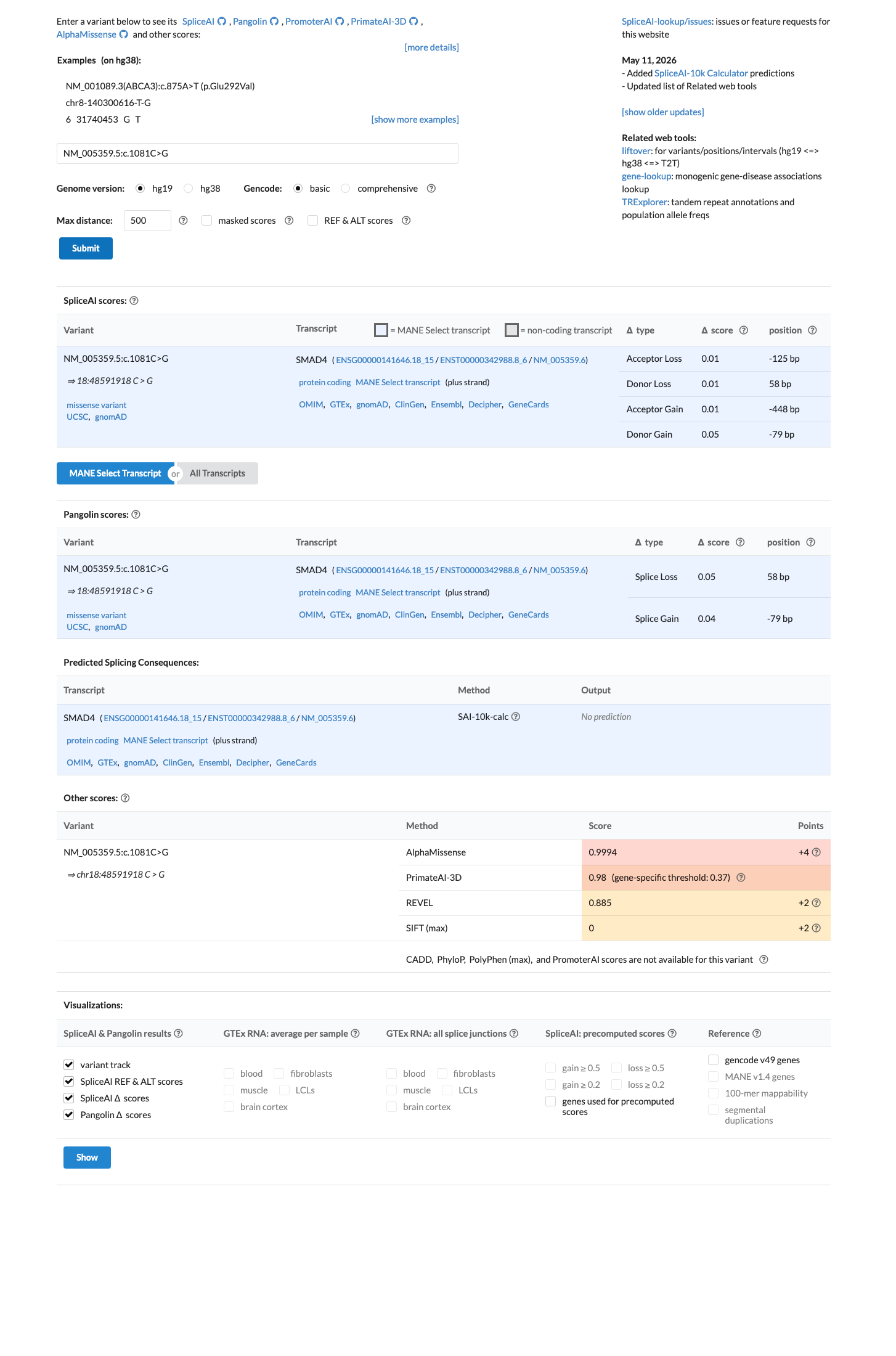
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.05). REVEL score = 0.885. BayesDel score = 0.512189.
Functional
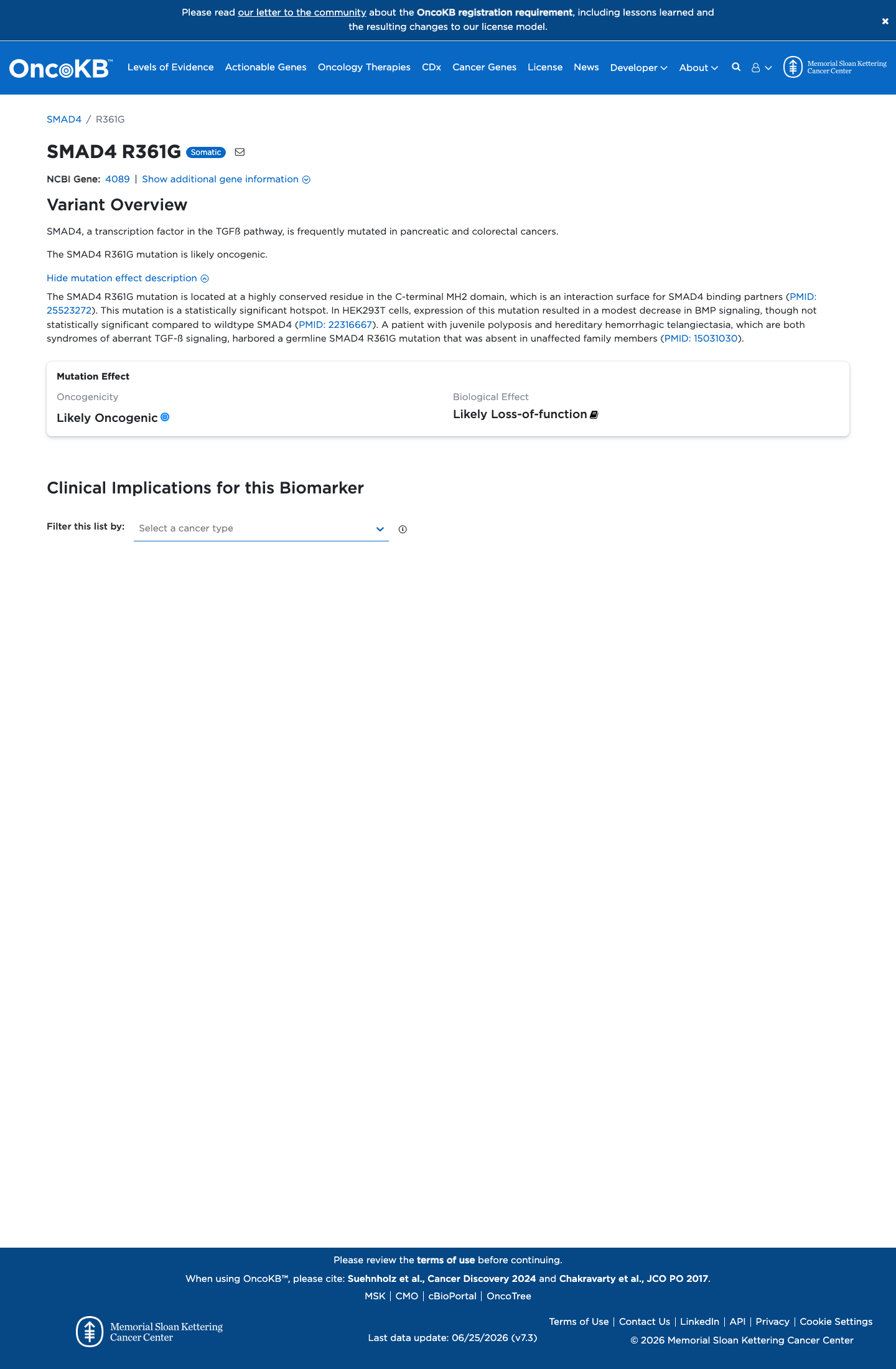
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.
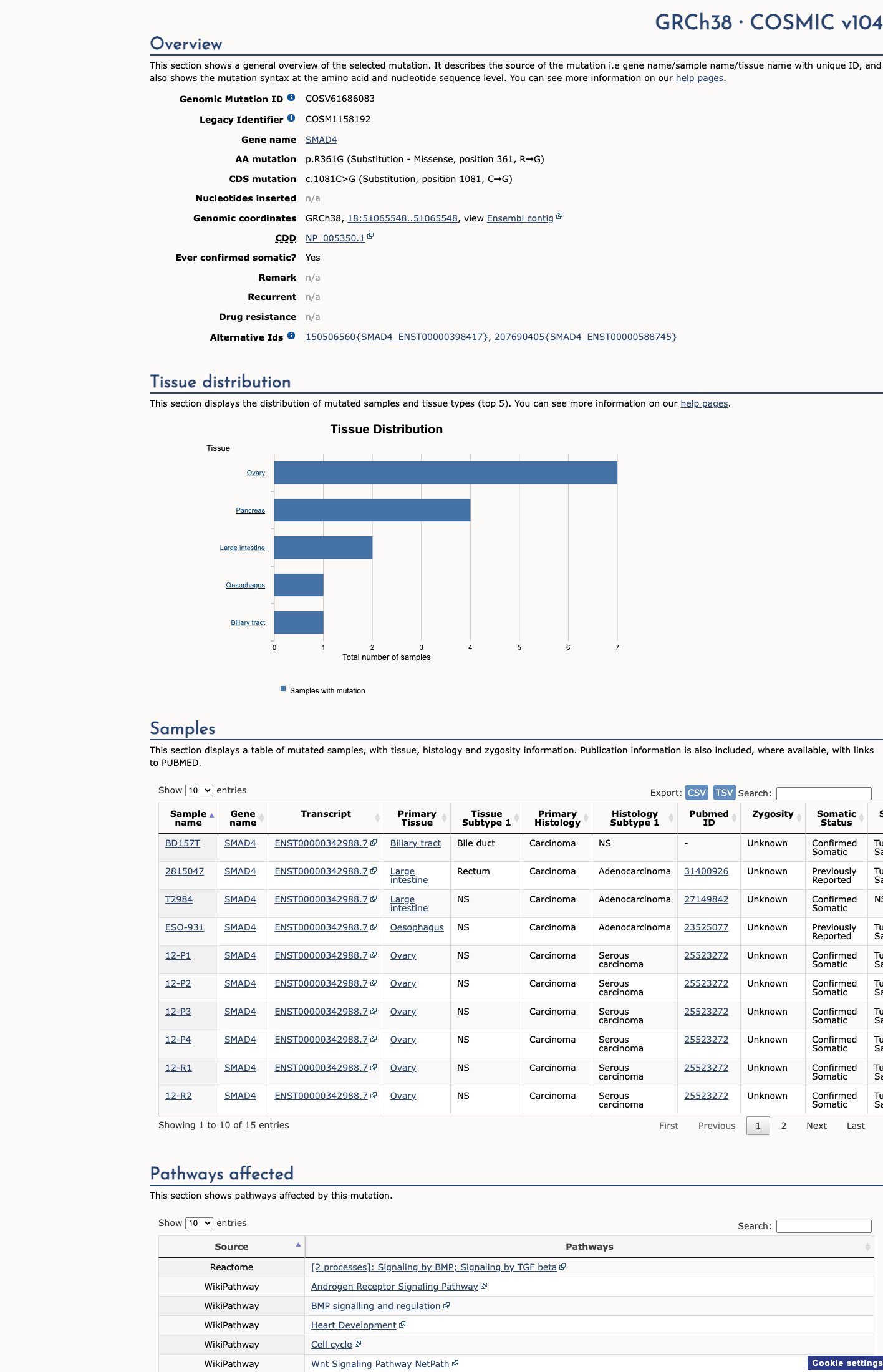
COSMIC
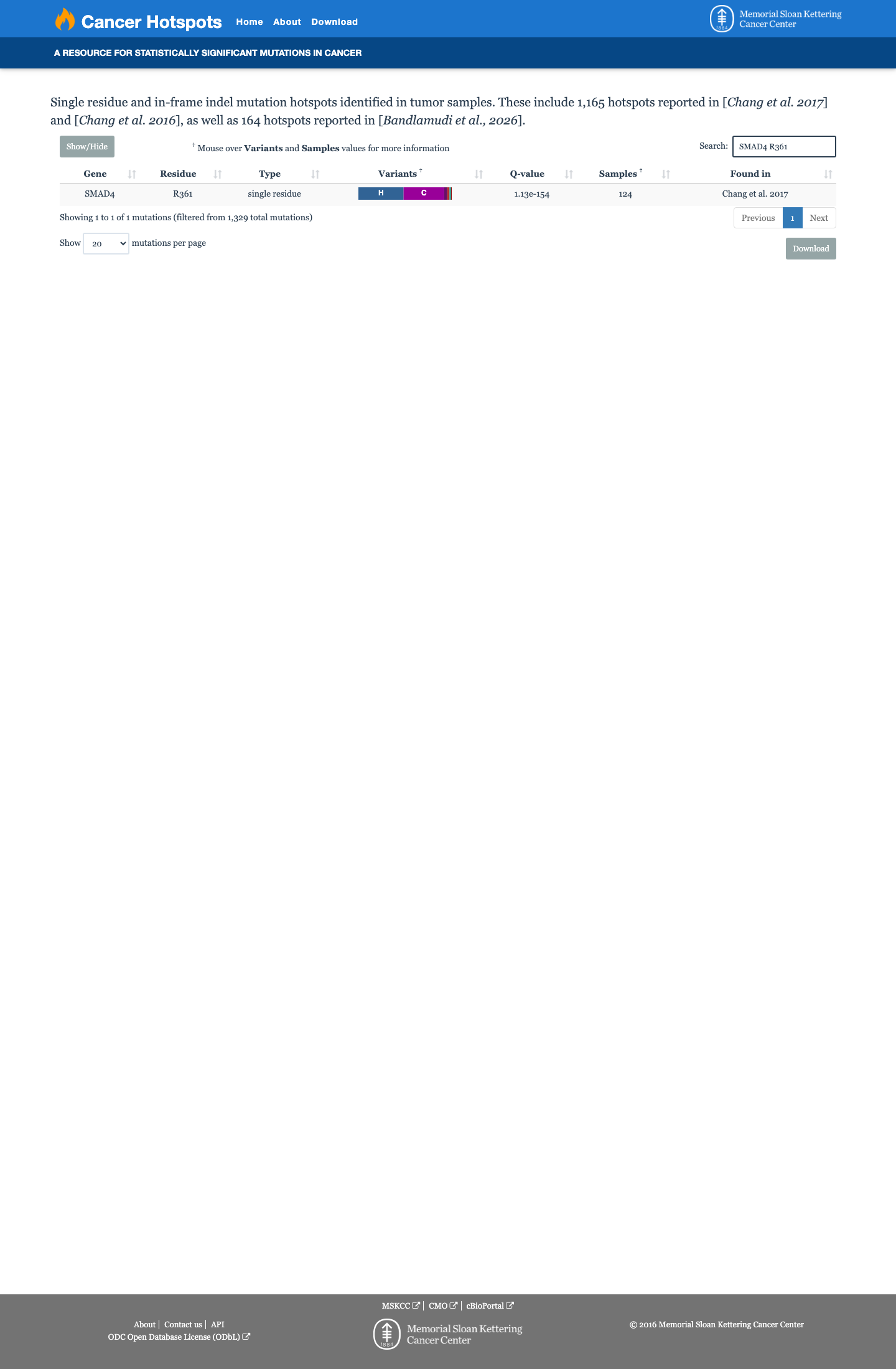
Cancer hotspots
Somatic evidence
Hotspot
COSMIC
This variant lies in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV61686083, n = 15 times).
Hotspots
This variant lies in a statistically significant hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 6 further PMIDs triaged but not cited — see Sources & References.
A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4).
Searched
1081C1081C>GR361GArg361Glyc.1081
Found
c.1081C>G (R361G) identified as a de novo mutation in the proband of Family 230 with juvenile polyposis and hereditary hemorrhagic telangiectasia overlap syndrome. The proband presented with colonic juvenile polyps, anemia, telangiectases, and epistaxis. Mutation was absent from both unaffected parents and chromosome 18 haplotypes confirmed biological parentage. Mutation cosegregated with the JP-HHT phenotype where multiple affected family members were available.
Variant
✓ Names this variant — characterised directly
Applied to
→PP4 supports · met
→PS2 supports · met
→PS4 supports · met
Why
Confirmed de novo variant in JP-HHT overlap syndrome; provides PS2 (strong), PS4 (moderate), and PP4 (supporting) evidence.
The third de-novo mutation was found in family 230; the mutation (1081C→G, R361G) was seen only in the proband who has many colonic polyps and both telangiectases and epistaxis. The mutation was not found in either unaffected parent.
Location Table 1 (line 814); Results text (lines 912-916); Figure (Family 230 pedigree, lines 420-421) · Context Clinical ascertainment, Sanger sequencing of MADH4 coding exons, chromosome 18 haplotype analysis for parentage confirmation · full text
The rate of germline mutations and large deletions of SMAD4 and BMPR1A in juvenile polyposis.
Searched
1081C1081C.GR361GArg361Glyc.1081
Found
c.1081C>G (R361G) listed in Table 1 among SMAD4 mutations identified by direct sequencing in 102 juvenile polyposis probands. Proband JP71 carried this variant as a sporadic case with one affected family member. The variant falls within MRR1 (Mutational Rich Region 1), the most frequently mutated region of SMAD4 (25% of all reported mutations located in codons 330-370 of the MH2 domain).
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
→PS4 supports · met
Why
Variant identified in an independent sporadic JPS proband; confirms location in critical MH2 domain hotspot (MRR1). Supports PS4 and PM1.
1081C.G / R361G
Location Table 1 (lines 231-233); Discussion (lines 366-388) · Context Direct Sanger sequencing of SMAD4 coding exons and intron-exon boundaries in 102 JPS probands · full text
Sources & reference links
Triaged references · 6 PMIDs not cited in assessment
10751092 ↗
Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome).
CLINVAR
15235019 ↗
The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR