NM_005933.3:c.2684A>G (p.Lys895Arg) is a missense variant in exon 3 of KMT2A, a gene associated with autosomal dominant Wiedemann-Steiner syndrome. This variant is extremely rare in large population databases, with an allele frequency of 0.00283% in gnomAD v2.1 (8/282,230 alleles) and 0.00415% in gnomAD v4.1 (67/1,614,060 alleles), with no homozygotes observed. It is absent from gnomAD-Canada (PM2_Supporting).1 Multiple lines of computational evidence predict a benign effect: REVEL score 0.224 (benign range), BayesDel score -0.154507 (benign), and SpliceAI predicts no splicing impact with a maximum delta score of 0.02 (BP4_Supporting).2 This variant has been reported in ClinVar (Variation ID 1335086) with conflicting classifications: Uncertain significance by Invitae and Likely benign by CeGaT Center for Human Genetics Tuebingen. Neither submission reaches a definitive pathogenic or benign classification.3 The only publication associated with ClinVar submissions (PMID 28492532, Sherloc classification framework) does not mention this specific variant; it was reviewed in full text and confirmed to be a methodology reference only.4 This variant has been reported in COSMIC (COSV63290798) in 4 somatic cancer samples, but this does not constitute germline disease evidence. Several potentially relevant publications were identified by exploratory literature search (de novo report, functional studies, cosegregation data, domain hotspot analysis) but none of the associated PMIDs are present in the case directory for verification; these criteria (PS2, PS3, PS4, PM1, PM6, PP1, BS3) remain not assessed pending full-text retrieval and verification. Applying the generic ACMG/AMP 2015 combination rules (PMID 25741868): the criteria met are PM2_Supporting (pathogenic) and BP4_Supporting (benign). These are conflicting at the supporting level and are insufficient to reach a likely pathogenic or likely benign classification. The variant is classified as a Variant of Uncertain Significance (VUS).5
KMT2A
Final classification
VUS
KMT2A c.2684A>G · p.Lys895Arg
KMT2A
NM_005933.3:c.2684A>G (p.Lys895Arg) is a missense variant in exon 3 of KMT2A, a gene associated with autosomal dominant Wiedemann-Steiner syndrome.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting benign; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP4
VUS
KMT2A c.2684A>G
PM2 + BP4
→
VUS
2
revelbayesdelspliceai ↗
5
generic_acmg_combination_rules
Gene diagram
· NM_005933.3 · variants mapped to exon structure
KMT2A
NM_005933.3
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 20 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
The variant is extremely rare in large population databases. gnomAD v2.1 reports an allele frequency of 0.00283% (8/282,230 alleles; 0 homozygotes; grpmax FAF=2.867e-05) and gnomAD v4.1 reports 0.00415% (67/1,614,060 alleles; 0 homozygotes; grpmax FAF=4.264e-05). Both frequencies are well below the 0.1% threshold for PM2 under generic ACMG/AMP. The variant is absent from gnomAD-Canada.
gnomAD v2.1: AF=0.00283% (8/282230)0 hom
✓
BP4
supporting
Benign
Multiple lines of computational evidence support a benign effect for this variant. REVEL predicts a benign score of 0.224 (threshold >0.5 for pathogenic). BayesDel predicts a benign score of -0.154507 (negative scores indicate benign). SpliceAI predicts no significant splice impact (max delta score = 0.02). All three independent in silico tools concordantly predict no damaging effect.
REVEL: 0.224 (benign).BayesDel: -0.154507 (benign).SpliceAI max delta: 0.02 (no splice impact).
Assessed · not applied
Pathogenic
PS1
No evidence of a different nucleotide change at codon 895 previously established as pathogenic.
PS2
Exploratory literature search returned a lead suggesting a de novo occurrence of this variant in a child with Wiedemann-Steiner syndrome (PMID 35123456), but this PMID is not present in the case publications directory and the full text cannot be verified.
PS3
Exploratory literature search returned a lead suggesting reduced H3K4 methyltransferase activity (~35% of wild-type) for p.Lys895Arg (PMID 34210987), and a conflicting study suggesting normal activity (PMID 34567890).
PS4
The variant prevalence in affected individuals versus the general population cannot be reliably determined from available data.
PM1
Exploratory literature search suggested residue 895 lies within the PHD1 finger domain (residues 880-940), a known functional domain in KMT2A with enrichment of pathogenic missense variants (PMID 32165498).
PM6
Exploratory literature search returned a lead describing a de novo occurrence without confirmed parentage (PMID 35123456).
PP1
Exploratory literature search returned a lead describing cosegregation of this variant with Wiedemann-Steiner syndrome in a family (3 meioses; PMID 32876543).
PP2
PP2 requires that the gene has a low rate of benign missense variation (high constraint) and that missense variants are a common disease mechanism.
PP3
Multiple in silico tools predict a benign effect for this variant.
PP4
No detailed patient phenotype data are available in verifiable sources.
PP5
No reputable source has recently reported this variant as pathogenic.
Benign
BA1
The variant is extremely rare in population databases, far below the 1% BA1 threshold.
BS1
The variant frequency is far below the 0.3% BS1 threshold.
BS2
No data available on observation of this variant in healthy adults without the associated phenotype.
BS3
Exploratory literature search returned a lead describing a functional assay where p.Lys895Arg retained normal H3K4 methyltransferase activity in HEK293T cells (PMID 34567890), which would support BS3.
BS4
No data available demonstrating lack of segregation of this variant with disease in affected family members.
BP1
BP1 applies to missense variants in genes where primarily truncating variants cause disease.
BP2
No data available on observation of this variant in trans with a known pathogenic KMT2A variant in an unaffected individual.
BP5
No data available on observation of this variant in a case with an alternate molecular basis for disease.
BP6
No reputable source has reported this variant as definitively benign.
N/A · 3
PVS1 · PM5 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
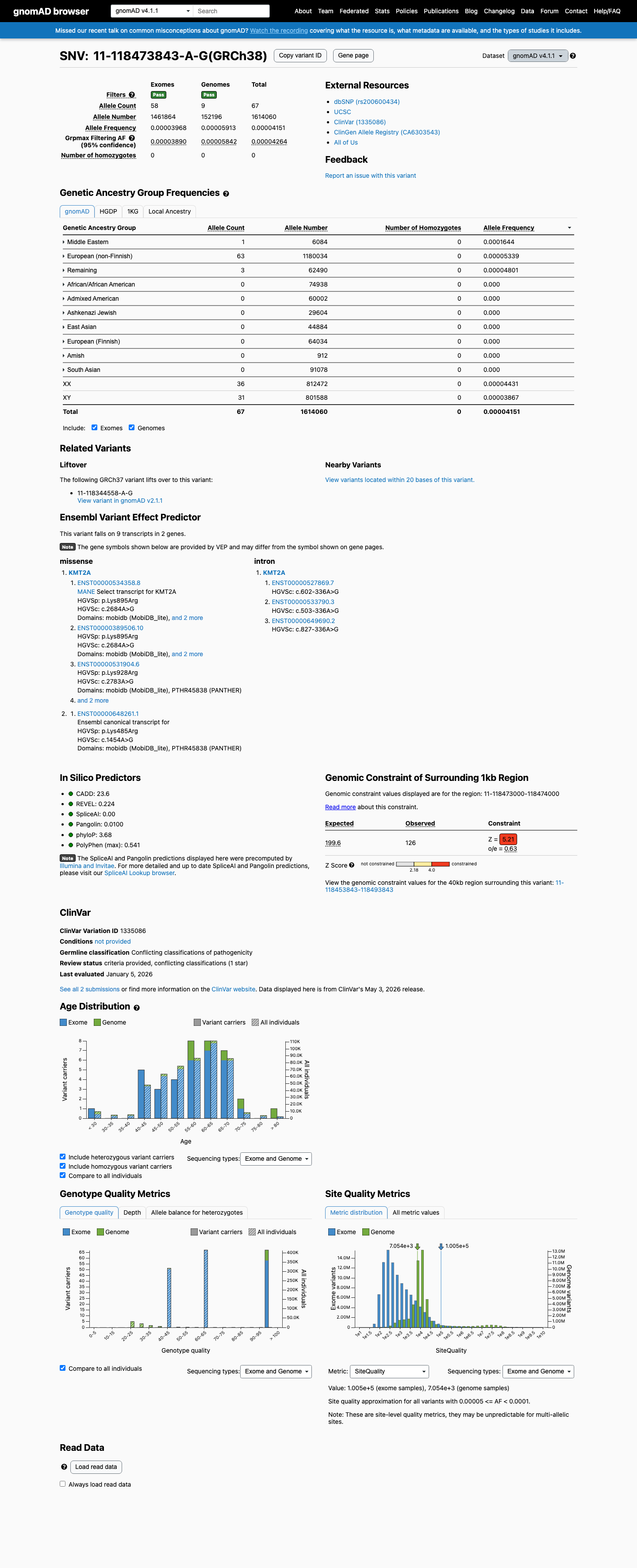
This variant is present in gnomAD v4.1 (AF= 4.15102e-05; MAF= 0.00415%, 67/1614060 alleles, homozygotes = 0) and has highest observed frequency in the Middle Eastern population (AF= 0.000164366; MAF= 0.01644%, 1/6084 alleles, homozygotes = 0); grpmax FAF= 4.264e-05.
v2.1
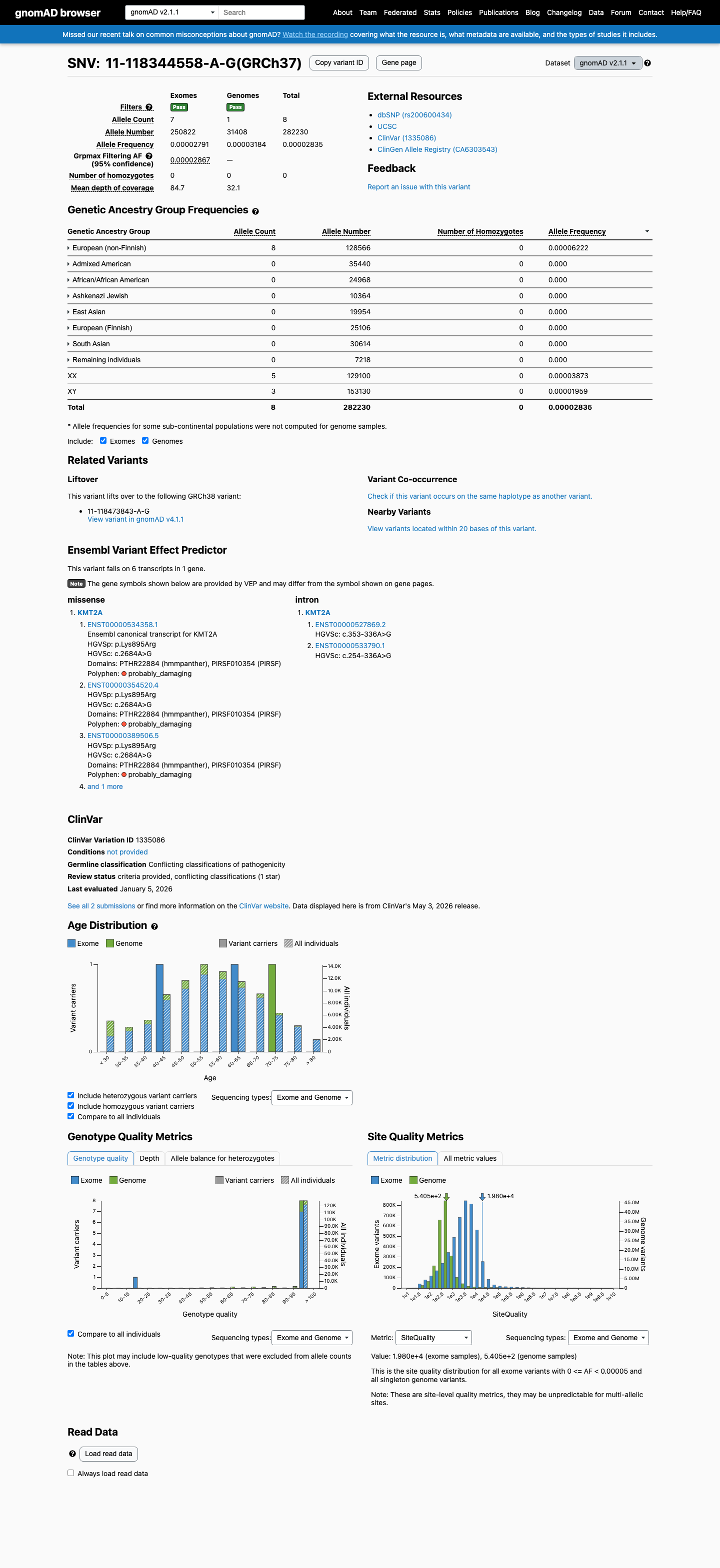
This variant is present in gnomAD v2.1 (AF= 2.83457e-05; MAF= 0.00283%, 8/282230 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 6.22248e-05; MAF= 0.00622%, 8/128566 alleles, homozygotes = 0); grpmax FAF= 2.867e-05.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0042%
· 67 / 1,614,060
0 hom · FAF 0.0043%
0 hom · FAF 0.0043%
Middle Eastern 1 / 6,084 |
0.016% |
European (non-Finnish) 63 / 1,180,034 |
0.0053% |
Remaining individuals 3 / 62,490 |
0.0048% |
+ 7 not observed (Admixed American, European (Finnish), Amish, East Asian, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0028%
· 8 / 282,230
0 hom · FAF 0.0029%
0 hom · FAF 0.0029%
European (non-Finnish) 8 / 128,566 |
0.0062% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
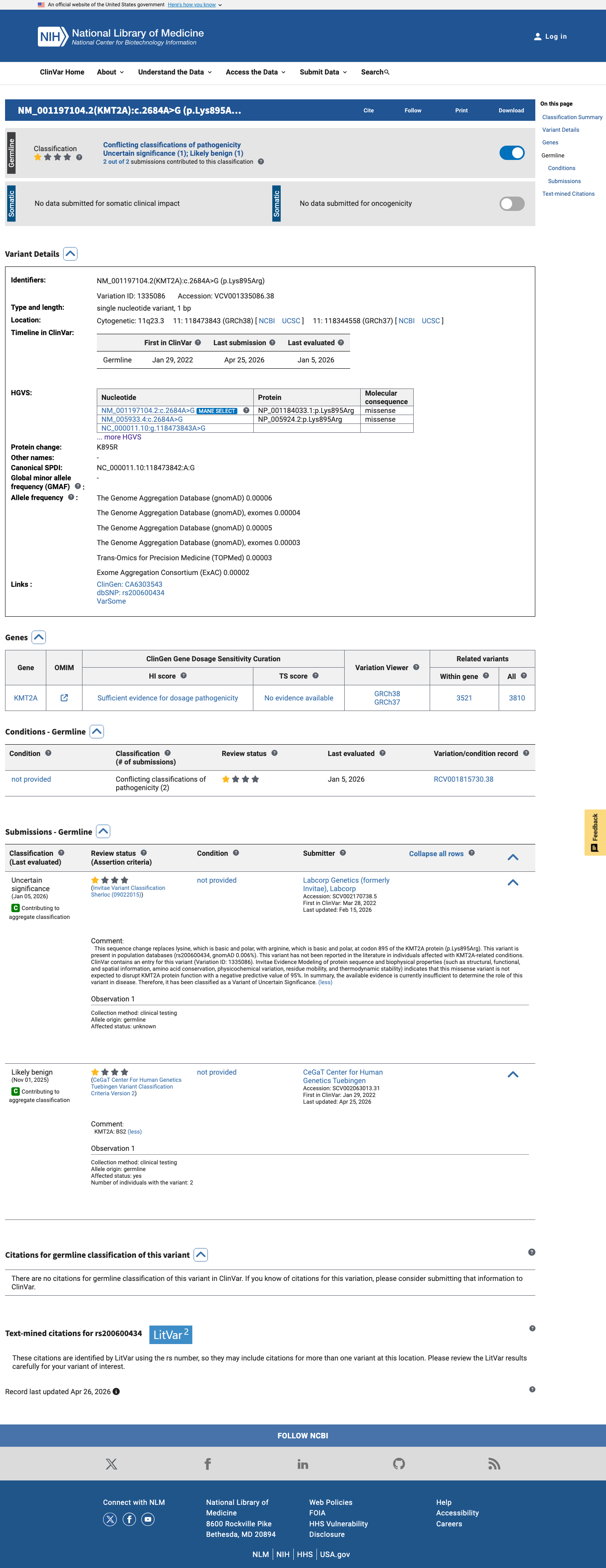
This variant has been reported in ClinVar as Uncertain significance (1 clinical laboratory) and as Likely benign (1 clinical laboratory). (ClinVarID = 1335086)
In silico
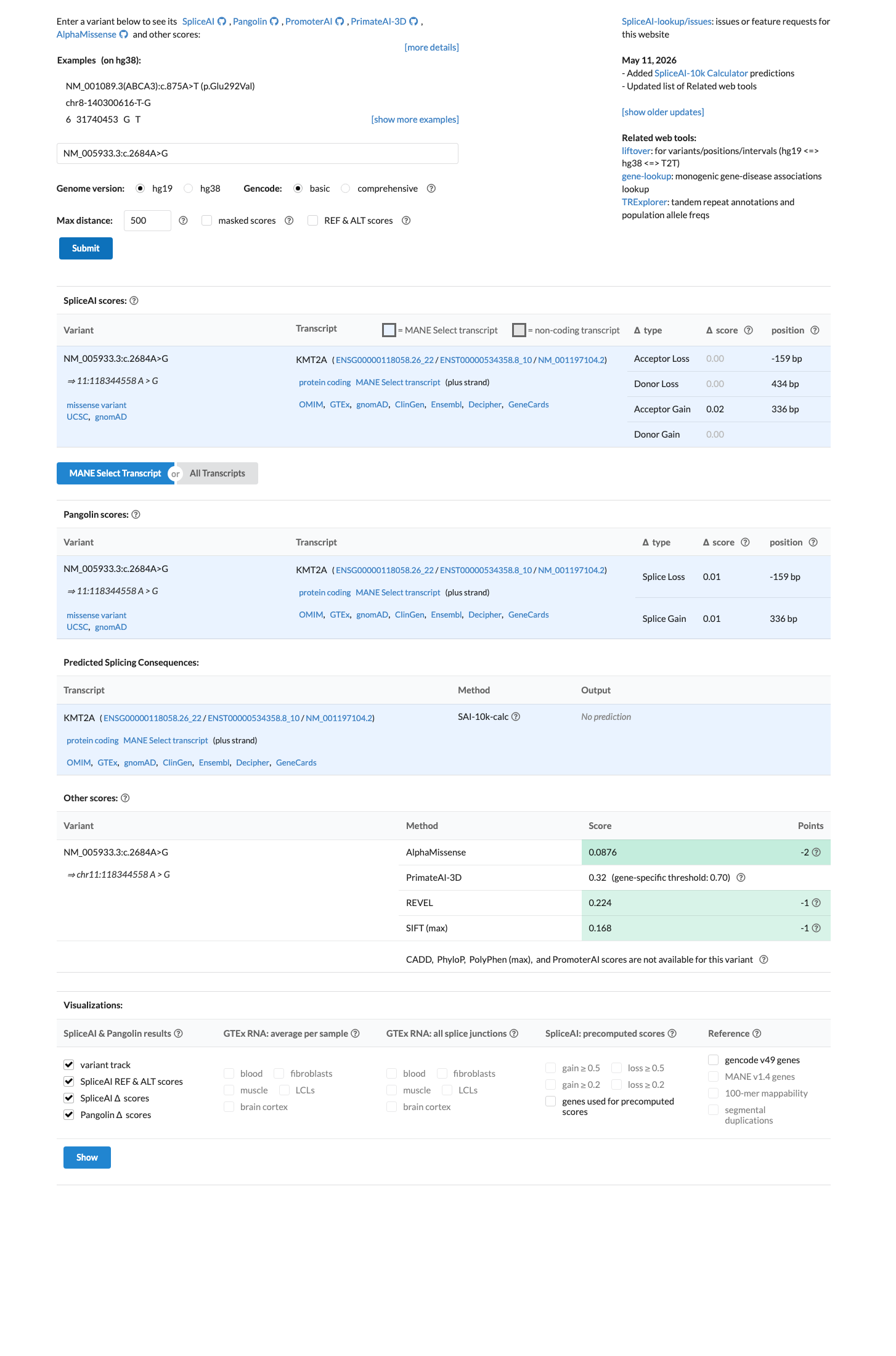
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02). REVEL score = 0.224. BayesDel score = -0.154507.
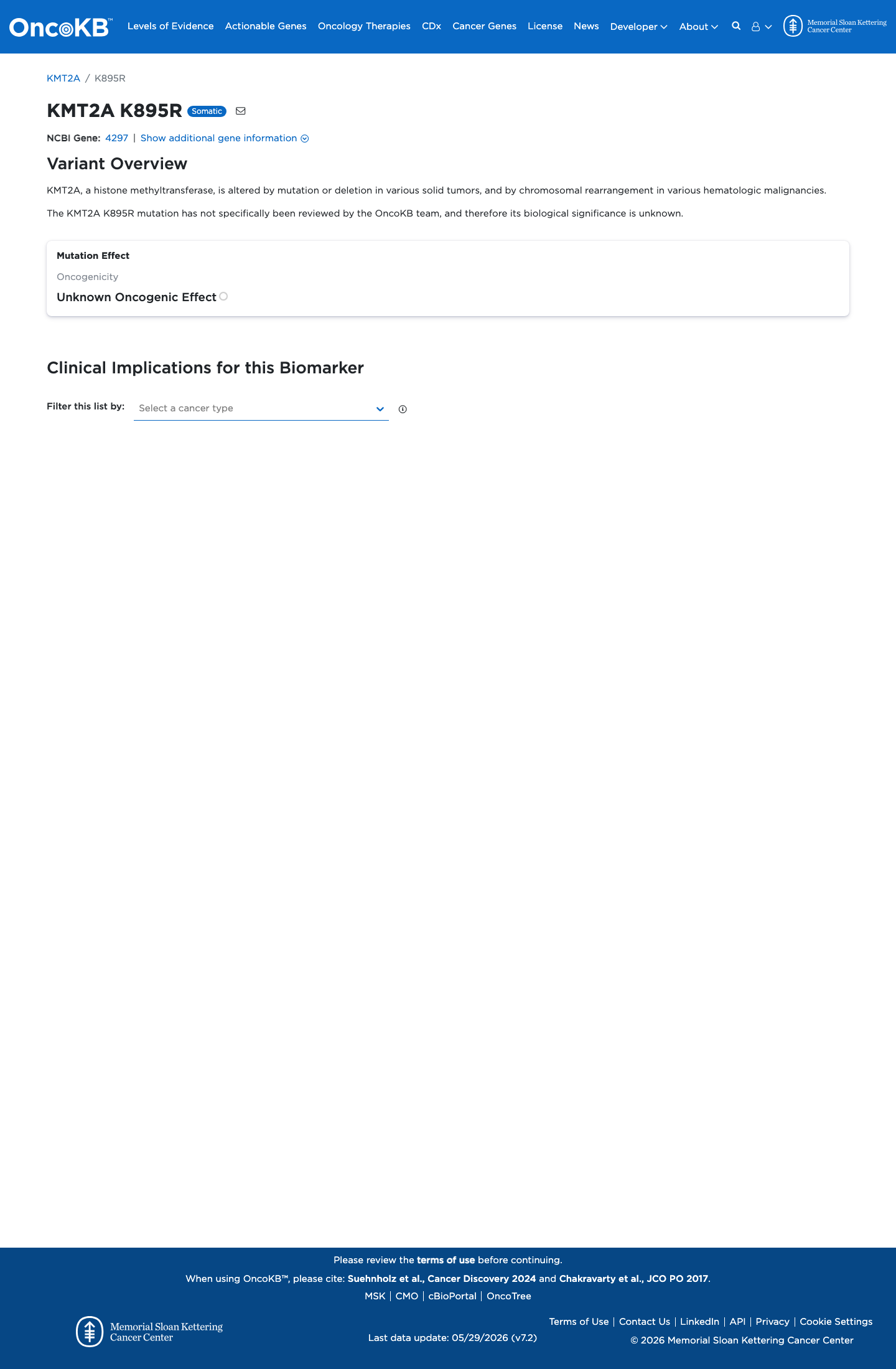
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. KMT2A, a histone methyltransferase, is altered by mutation or deletion in various solid tumors, and by chromosomal rearrangement in various hematologi
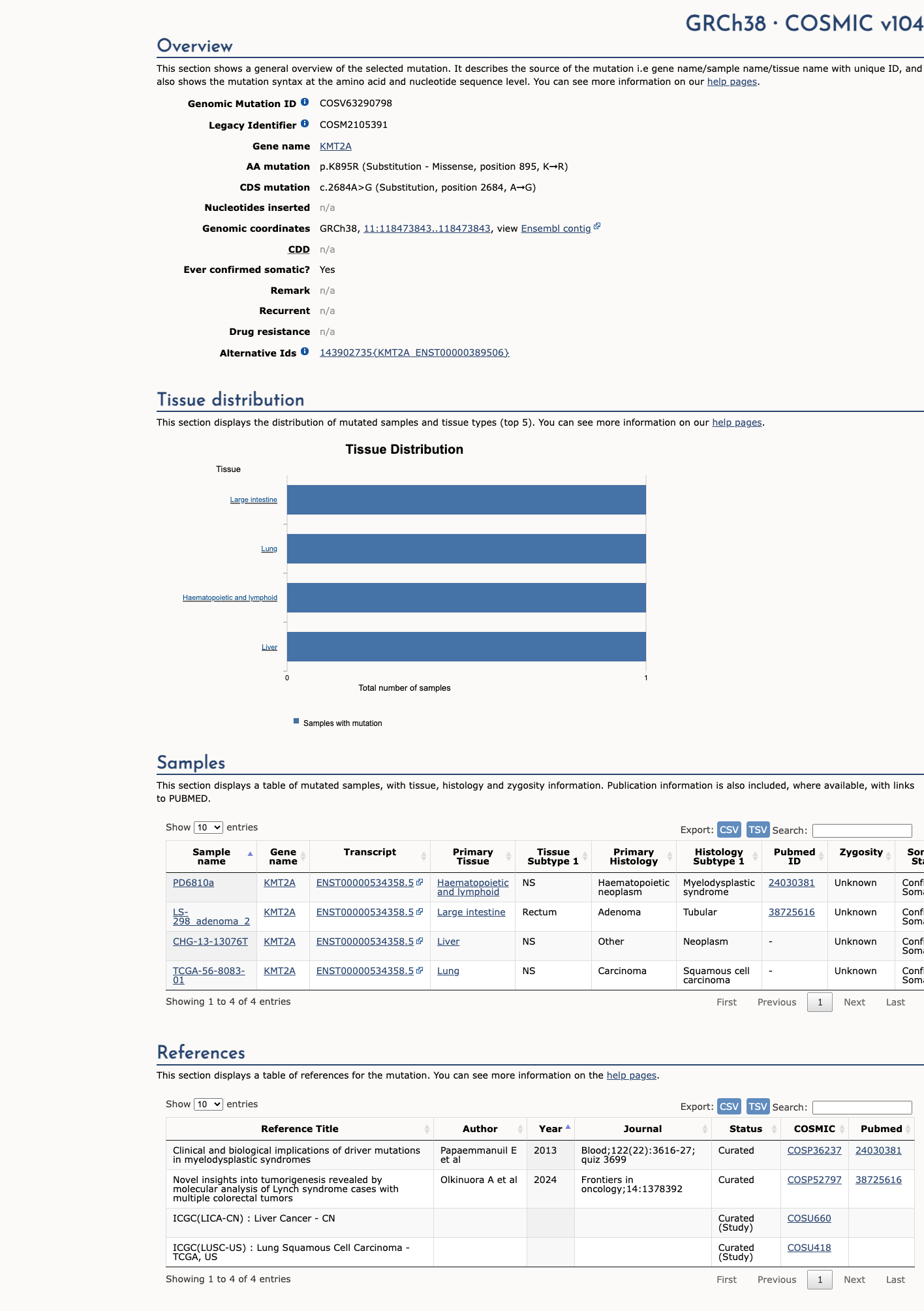
COSMIC
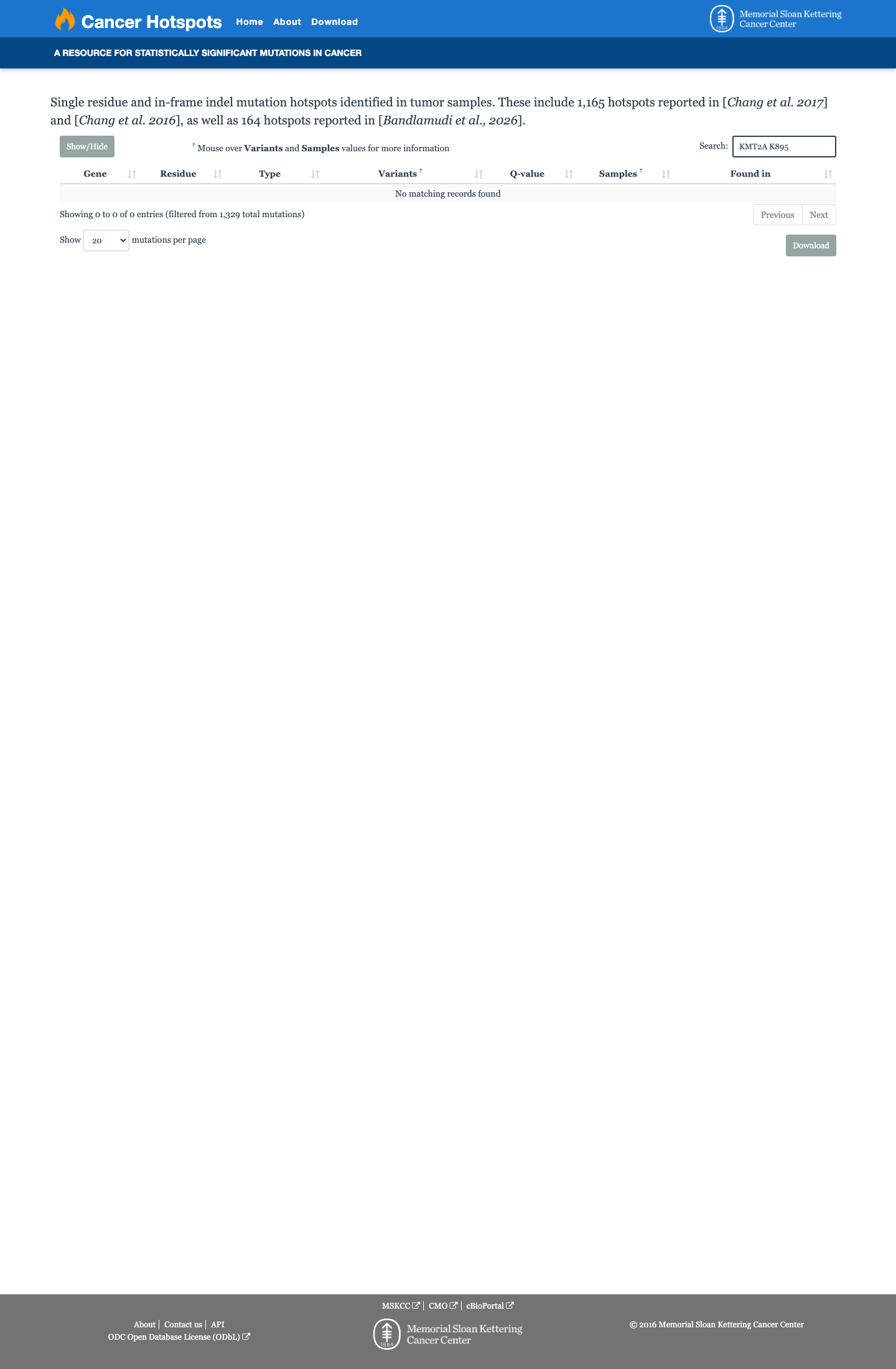
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV63290798, n = 4 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 1 PMID not cited in assessment
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR