Classification rationale
VUS
KMT2A c.8956G>A
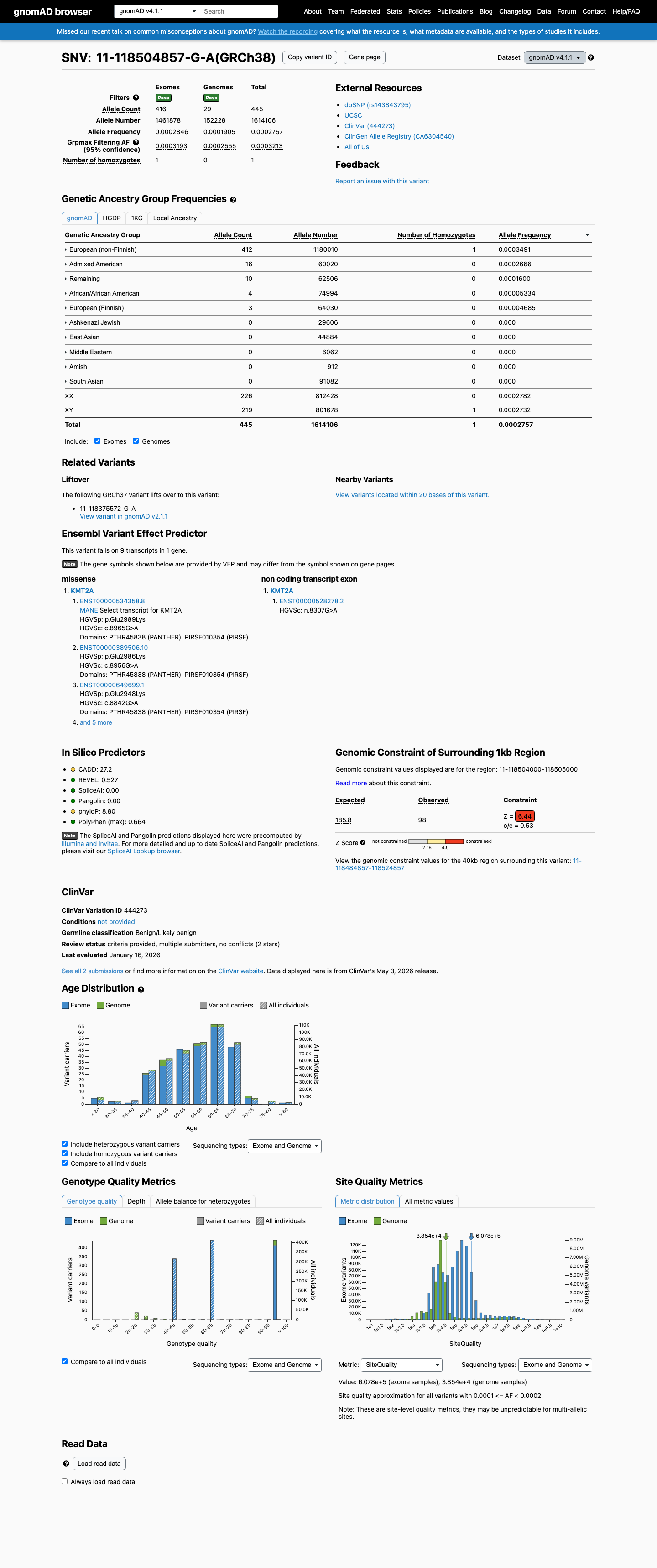
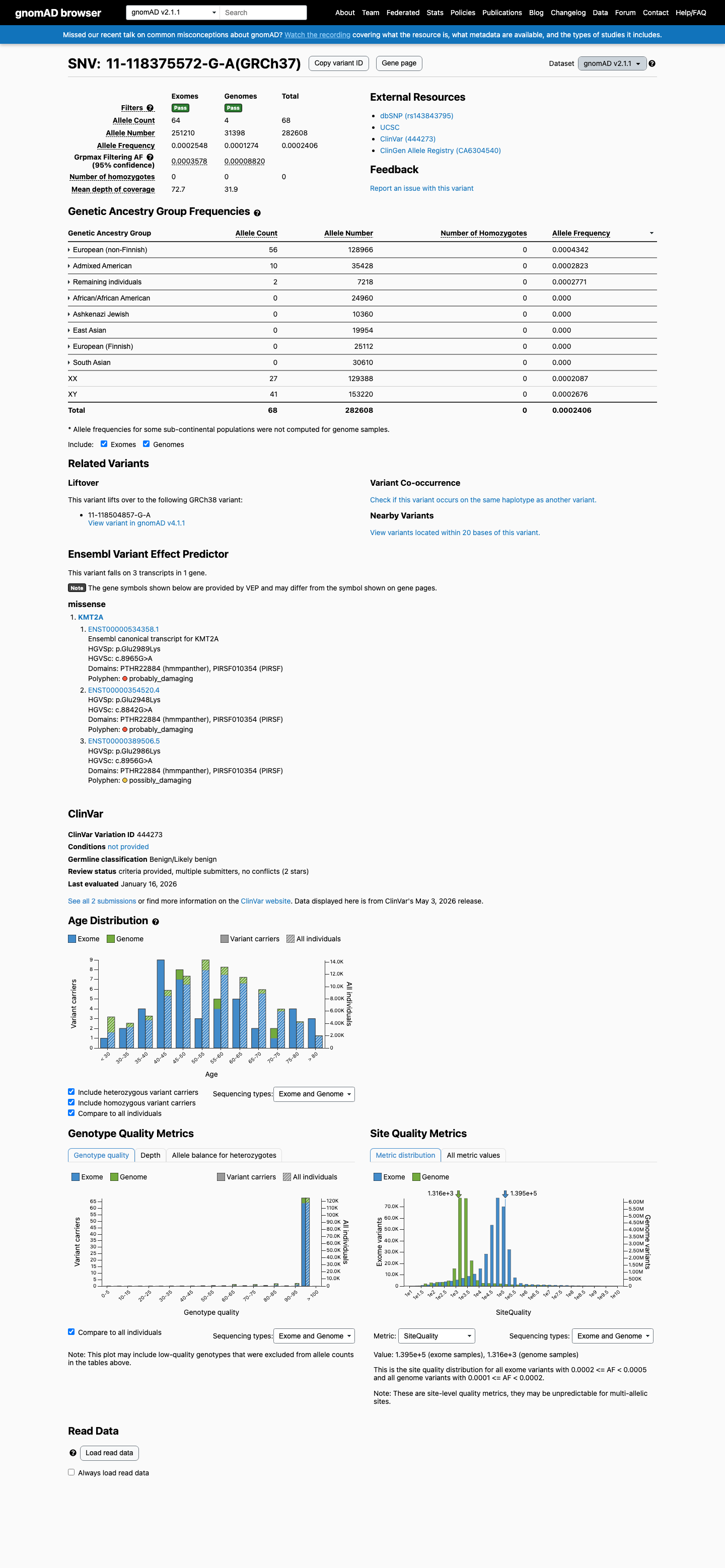
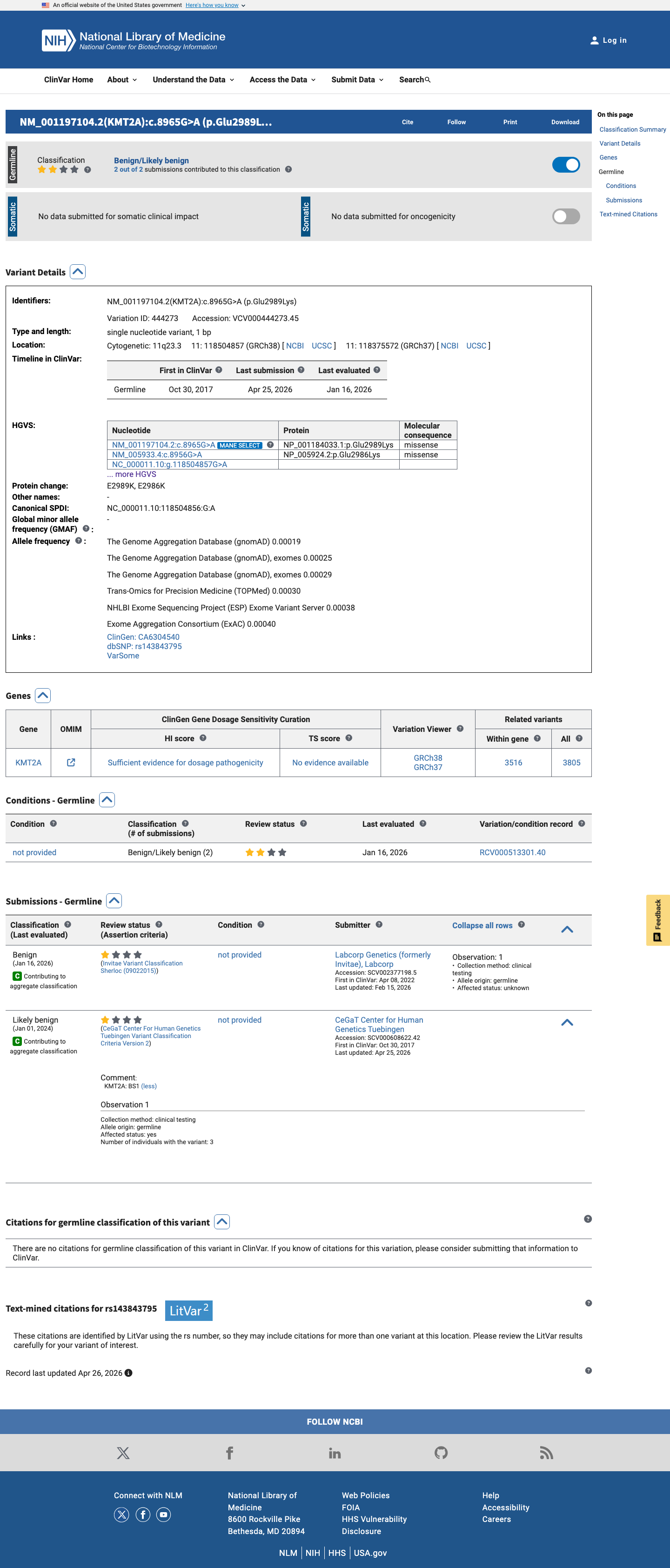
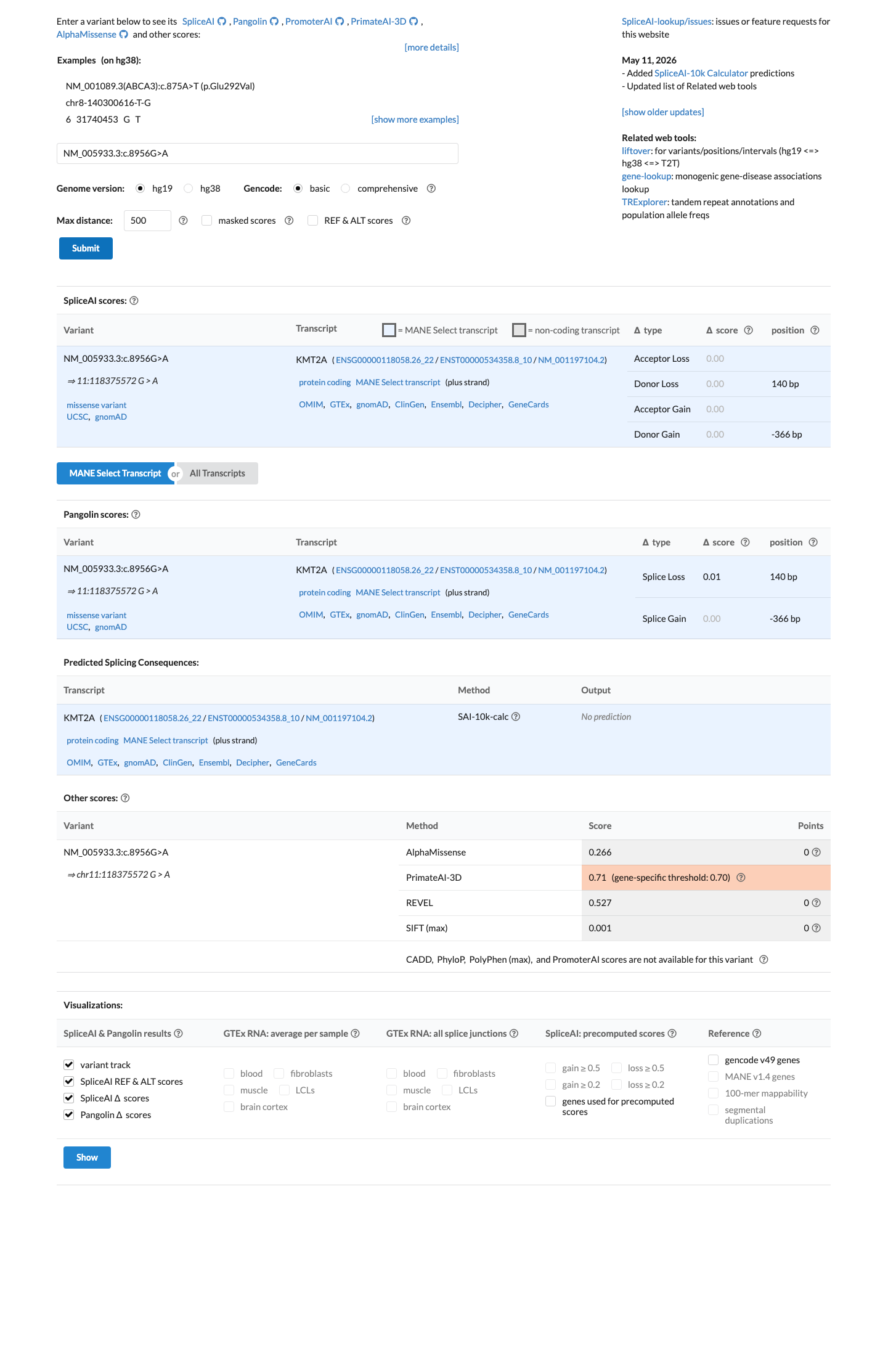
The KMT2A c.8956G>A (p.Glu2986Lys) variant has been reported in ClinVar with benign and likely benign single-submitter classifications.1 This variant is present in population databases, with gnomAD v2.1 AF 0.02406% (68/282608) and gnomAD v4.1 AF 0.02757% (445/1614106), including 1 homozygote in gnomAD v4.1.2 Available hotspot review did not identify this residue within a statistically significant mutational hotspot.3 In silico results are inconclusive overall: SpliceAI predicts no significant splice effect (max delta score 0.00), while REVEL 0.527 and BayesDel 0.269249 do not provide a concordant benign or pathogenic signal.4