NM_006231.4:c.-11C>T is a non-coding promoter/5'UTR substitution 11 bases upstream of the ATG start codon. It is present at extremely low frequency in gnomAD v4.1 (3/1,498,064 alleles, AF 0.00020%, all South Asian) and absent from gnomAD v2.1 and gnomAD-Canada, meeting PM2 at supporting level.1 PVS1 does not apply as this is a non-coding variant and not a null variant. The León-Castillo 2020 custom POLE framework criteria (PM1, PS4, PP3, BP4) apply only to exonuclease-domain missense variants and do not extend to this promoter variant.2 SpliceAI predicts no splice impact (max delta 0.00). No functional studies, segregation data, de novo reports, case-control data, or ClinVar classifications are available for this variant.3 No publications specifically mention NM_006231.4:c.-11C>T. Five papers reviewed in the PVS1 gene context support POLE loss-of-function as a germline disease mechanism at the gene level but do not report this specific variant. With only PM2_Supporting met, this variant does not meet any ACMG/AMP 2015 classification threshold (requires ≥2 pathogenic supporting criteria with additional evidence, or BS1/BA1 thresholds for benign classification). The variant is classified as a Variant of Uncertain Significance (VUS).4
POLE
Final classification
VUS
POLE c.-11C>T · p.?
POLE
NM_006231.4:c.-11C>T is a non-coding promoter/5'UTR substitution 11 bases upstream of the ATG start codon. It is present at extremely low frequency in gnomAD v4.1 (3/1,498,064 alleles, AF 0.00020%, all South Asian) and absent from gnomAD v2.1 and gnomAD-Canada, meeting PM2 at supporting level.
Only PM2 is met at supporting strength (gnomAD v4.1: 3/1,498,064 alleles, AF 0.00020%). All other criteria are not_met or not_applicable. Under the Leó-Castillo 2020 custom POLE framework combination rules (which mirror standard ACMG/AMP 2015 thresholds), a single supporting criterion does not satisfy any classification tier: Pathogenic requires at minimum PVS1+2Supporting or 2Strong; Likely Pathogenic requires at minimum PVS1+1Moderate or 1Strong+2Supporting or 3Moderate; Benign requires BA1 or 2 Strong benign; Likely Benign requires 1 Strong benign + 1 Supporting benign or 2 Supporting benign. With only PM2_Supporting met and no benign criteria, the variant defaults to Variant of Uncertain Significance.
Classification rationale
PM2
VUS
POLE c.-11C>T
PM2
→
VUS
2
pvs1_variant_assessmentvcep_path_250_323
4
generic_acmg_combination_rules
Gene diagram
· NM_006231.4 · variants mapped to exon structure
POLE
NM_006231.4
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 18 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_006231.4:c.-11C>T is present at extremely low frequency in population databases. gnomAD v4.1 reports 3 alleles out of 1,498,064 (AF 2.0e-06, 0.00020%), all in the South Asian subpopulation (SAS AF 3.75e-05). The variant is absent from gnomAD v2.1 and gnomAD-Canada. The total allele frequency is well below the 0.1% PM2 threshold, consistent with absence from controls at sufficient depth.
gnomAD v4.1: 3/1498064 alleles (AF 0.00020%
Assessed · not applied
Pathogenic
PVS1
NM_006231.4:c.-11C>T is a promoter/5'UTR substitution 11 bases upstream of the ATG start codon.
PS2
No de novo data available for NM_006231.4:c.-11C>T.
PS3
No functional studies are available for NM_006231.4:c.-11C>T.
PS4
The León-Castillo 2020 custom POLE PS4 framework applies only to exonuclease-domain missense variants recurrent in COSMIC/TCGA endometrial carcinoma cohorts.
PM1
The León-Castillo 2020 custom POLE PM1 framework assigns PM1 only to specific exonuclease-domain missense variants (e.g., P286R, V411L, S297F, A456P, S459F at strong; F367S, L424I at moderate; A465V, L424V at supporting).
PM6
No de novo evidence is available for NM_006231.4:c.-11C>T.
PP1
No segregation data are available for NM_006231.4:c.-11C>T.
PP3
The León-Castillo 2020 custom PP3 rule requires the variant to be a missense variant present in Supplementary Tables S2 or S3 with REVEL class 'likely disease causing' and ≤1 benign in silico result.
PP4
No phenotype specificity data are available.
PP5
No reputable source (ClinVar, expert panel, published report) has classified NM_006231.4:c.-11C>T as pathogenic.
Benign
BA1
gnomAD v4.1 allele frequency is 0.00020% (grpmax FAF 9.95e-06), well below the 1% BA1 threshold.
BS1
gnomAD v4.1 allele frequency is 0.00020%, below the 0.3% BS1 threshold.
BS2
No data on observation of NM_006231.4:c.-11C>T in healthy adults for a fully penetrant early-onset disorder.
BS3
No well-established functional studies demonstrate no deleterious effect of NM_006231.4:c.-11C>T.
BS4
No segregation data are available to assess lack of segregation with disease for NM_006231.4:c.-11C>T.
BP2
No evidence that NM_006231.4:c.-11C>T has been observed in trans with a pathogenic variant for a fully penetrant dominant disorder, or in cis with a pathogenic variant regardless of inheritance pattern.
BP4
The León-Castillo 2020 custom BP4 rule applies only to missense variants in Supplementary Tables S2/S3 with REVEL class 'likely benign' and ≥4 benign in silico results.
BP5
No evidence that NM_006231.4:c.-11C>T has been observed in a case with an alternative molecular cause for disease.
N/A · 7
PS1 · PM5 · PP2 · BP1 · BP3 · BP6 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
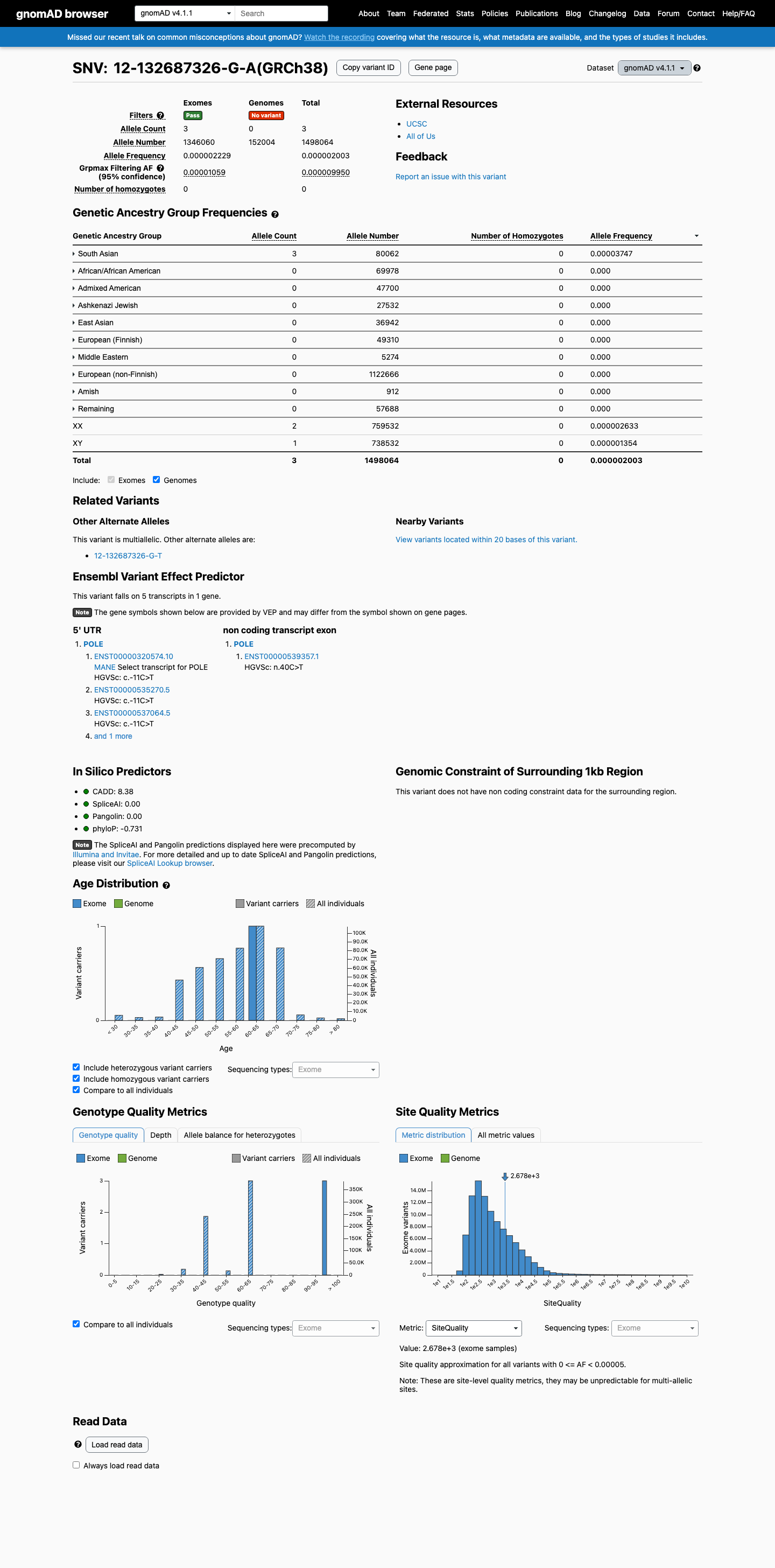
This variant is present in gnomAD v4.1 (AF= 2.00258e-06; MAF= 0.00020%, 3/1498064 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 3.7471e-05; MAF= 0.00375%, 3/80062 alleles, homozygotes = 0); grpmax FAF= 9.95e-06.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0002%
· 3 / 1,498,064
0 hom · FAF 0.001%
0 hom · FAF 0.001%
South Asian 3 / 80,062 |
0.0037% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish, African/African American, European (non-Finnish))
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links