NM_006231.4:c.2468+16_2468+21dup is an intronic duplication in intron 21 of POLE, located at positions +16 to +21 downstream of exon 21. This variant is absent from ClinVar and has not been reported in COSMIC or the published literature in association with disease.1 The variant is present in gnomAD at very low frequency (v2.1 AF = 0.0078%, v4.1 AF = 0.0079%) with no homozygotes observed, meeting PM2 at supporting strength.2 SpliceAI predicts no splicing impact (max delta = 0.02), meeting BP4 at supporting strength.3 The custom POLE framework criteria (PM1, PS4, PP3, BP4 via León-Castillo et al. 2020) are missense-specific and do not apply to this intronic duplication.4 PVS1 is not applicable as the variant is outside the canonical splice consensus and does not meet null-variant criteria per ClinGen PVS1 framework.5 With only PM2_Supporting and BP4_Supporting applied, evidence is insufficient to classify this variant as either pathogenic or benign. The variant is classified as a variant of uncertain significance (VUS).6
POLE
Final classification
VUS
POLE c.2468+16_2468+21dup · p.?
POLE
NM_006231.4:c.2468+16_2468+21dup is an intronic duplication in intron 21 of POLE, located at positions +16 to +21 downstream of exon 21.
The León-Castillo et al. 2020 custom POLE framework provides standard ACMG/AMP 2015 final combination rules. Only two criteria are met: PM2 at supporting strength (pathogenic) and BP4 at supporting strength (benign). This combination of one pathogenic supporting and one benign supporting criterion satisfies no pathogenic, likely pathogenic, benign, or likely benign rule in the framework. The conflicting evidence defaults to VUS per ACMG/AMP 2015.
Classification rationale
PM2
BP4
VUS
POLE c.2468+16_2468+21dup
PM2 + BP4
→
VUS
4
vcep_path_250_323
6
generic_acmg_combination_rules
Gene diagram
· NM_006231.4 · variants mapped to exon structure
POLE
NM_006231.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 16 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is absent from ClinVar and present in gnomAD at very low allele frequency: v2.1 AF = 7.82 × 10⁻⁵ (22/281,506 alleles) and v4.1 AF = 7.90 × 10⁻⁵ (127/1,608,390 alleles). Both frequencies are well below the 0.1% PM2 threshold for non-VCEP assessment. No homozygotes observed. PM2 is met at supporting strength.
gnomAD v2.1: AF = 0.0078% (22/281506 alleles0 homozygotes)
✓
BP4
supporting
Benign
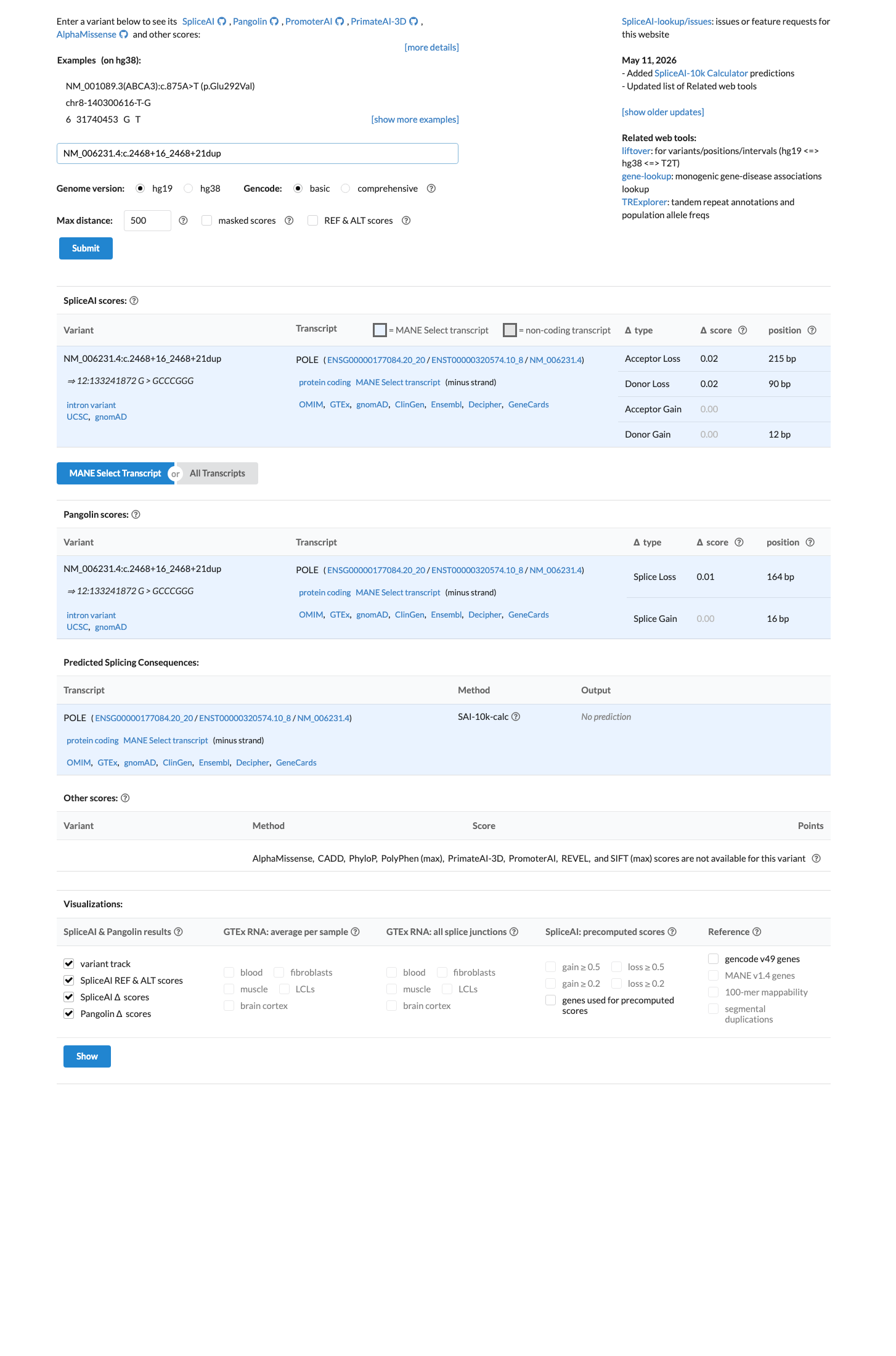
SpliceAI predicts no splicing impact for this intronic duplication (max delta score = 0.02, well below the 0.10 threshold). The variant lies at positions +16 to +21, outside the splice consensus region. The most pertinent computational tool for an intronic variant shows no evidence of altered splicing. REVEL and BayesDel are not applicable (not an SNV). BP4 is met at supporting strength based on SpliceAI prediction of no splicing impact.
SpliceAI max delta = 0.02 (no splicing impact predicted at acceptor or donor sites)SpliceAI scores: acceptor_gain=0.02all other delta scores <0.01
Assessed · not applied
Pathogenic
PS2
No de novo data available for this variant.
PS3
No functional studies available for this intronic duplication.
PS4
The variant is absent from ClinVar and COSMIC.
PM1
The custom POLE PM1 rules (from León-Castillo et al.
PM6
No de novo data available.
PP1
No segregation data available.
PP3
The custom POLE PP3 rule requires an exact missense variant in Supplementary Tables S2 or S3 with REVEL class 'likely disease causing' and ≤1 benign in silico result.
PP4
No phenotype or family history data are available for individuals carrying NM_006231.4:c.2468+16_2468+21dup.
PP5
The variant is absent from ClinVar.
Benign
BA1
gnomAD allele frequency is well below the 1% BA1 threshold.
BS1
gnomAD allele frequency is below the 0.3% BS1 threshold.
BS2
BS2 requires observation of the variant in a healthy adult individual for a fully penetrant disorder.
BS3
No functional studies demonstrating no deleterious effect are available for this intronic duplication.
BS4
No segregation data are available.
BP2
No data on in cis observation with a known pathogenic variant in POLE are available for this variant.
BP6
No reputable source has classified NM_006231.4:c.2468+16_2468+21dup as benign.
N/A · 10
PVS1 · PS1 · PM3 · PM4 · PM5 · PP2 · BP1 · BP3 · BP5 · BP7
Research & evidence
Population frequency

gnomAD v4.1

gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 7.89609e-05; MAF= 0.00790%, 127/1608390 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 0.000350795; MAF= 0.03508%, 21/59864 alleles, homozygotes = 0); grpmax FAF= 0.00023458.
v2.1
This variant is present in gnomAD v2.1 (AF= 7.81511e-05; MAF= 0.00782%, 22/281506 alleles, homozygotes = 0) and has highest observed frequency in the Remaining individuals population (AF= 0.000277855; MAF= 0.02779%, 2/7198 alleles, homozygotes = 0); grpmax FAF= 5.709e-05.
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0079%
· 127 / 1,608,390
0 hom · FAF 0.023%
0 hom · FAF 0.023%
Admixed American 21 / 59,864 |
0.035% |
African/African American 13 / 74,716 |
0.017% |
Remaining individuals 10 / 62,242 |
0.016% |
European (non-Finnish) 80 / 1,175,914 |
0.0068% |
East Asian 1 / 44,612 |
0.0022% |
South Asian 2 / 90,908 |
0.0022% |
+ 4 not observed (European (Finnish), Amish, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.0078%
· 22 / 281,506
0 hom · FAF 0.0057%
0 hom · FAF 0.0057%
Remaining individuals 2 / 7,198 |
0.028% |
Admixed American 5 / 35,350 |
0.014% |
African/African American 3 / 24,922 |
0.012% |
European (non-Finnish) 10 / 128,384 |
0.0078% |
East Asian 1 / 19,904 |
0.005% |
South Asian 1 / 30,584 |
0.0033% |
+ 2 not observed (Ashkenazi Jewish, European (Finnish))
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links