Classification rationale
BA1BP7
Benign
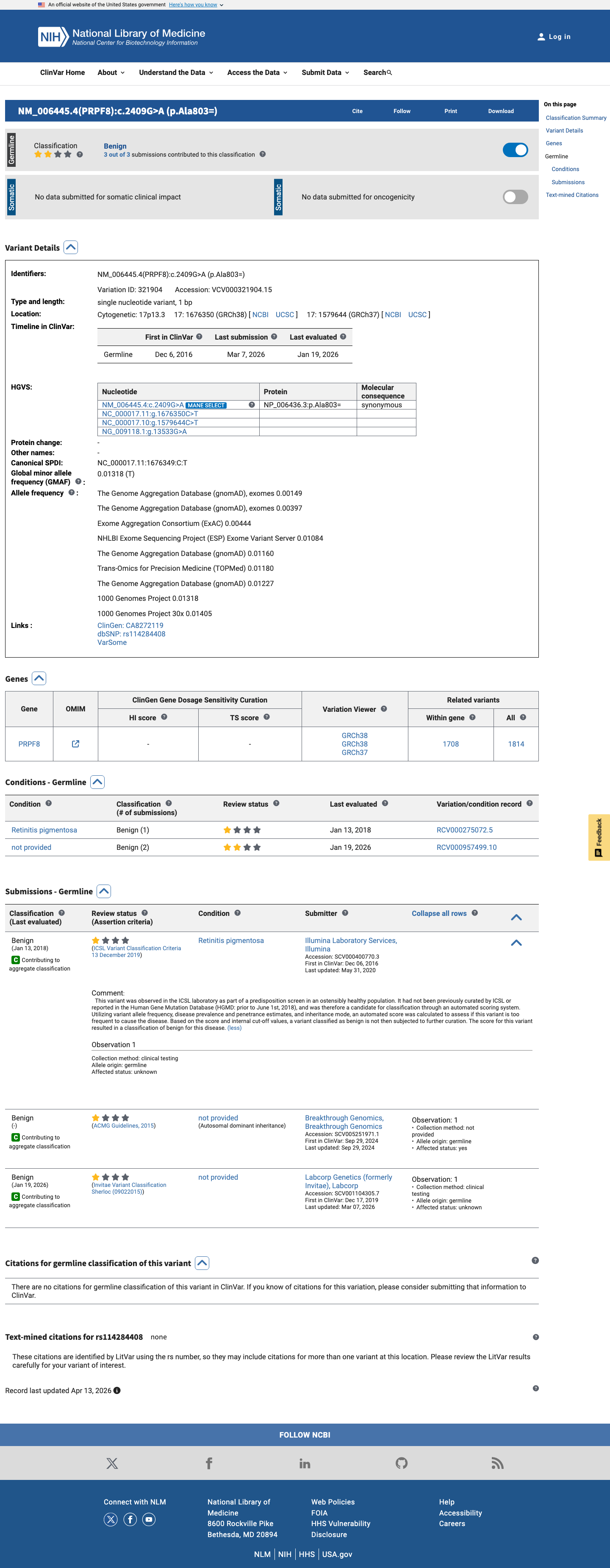
PRPF8 c.2409G>A
The PRPF8 NM_006445.3:c.2409G>A (p.Ala803=) variant has not been reported in ClinVar as a pathogenic germline variant and is listed there as benign.1 This variant is common in population databases, with a highest observed allele frequency of 3.582% in gnomAD v2.1, 3.636% in gnomAD v4.1, and 3.627% in gnomAD-Canada, which is above the 1% BA1 threshold.2 In silico splice prediction does not support an abnormal splicing effect, with a SpliceAI maximum delta score of 0.05.3
BA1 + BP7
→
Benign