Classification rationale
BP4BS1BA1BP7
Benign
LZTR1 c.2219+13C>T
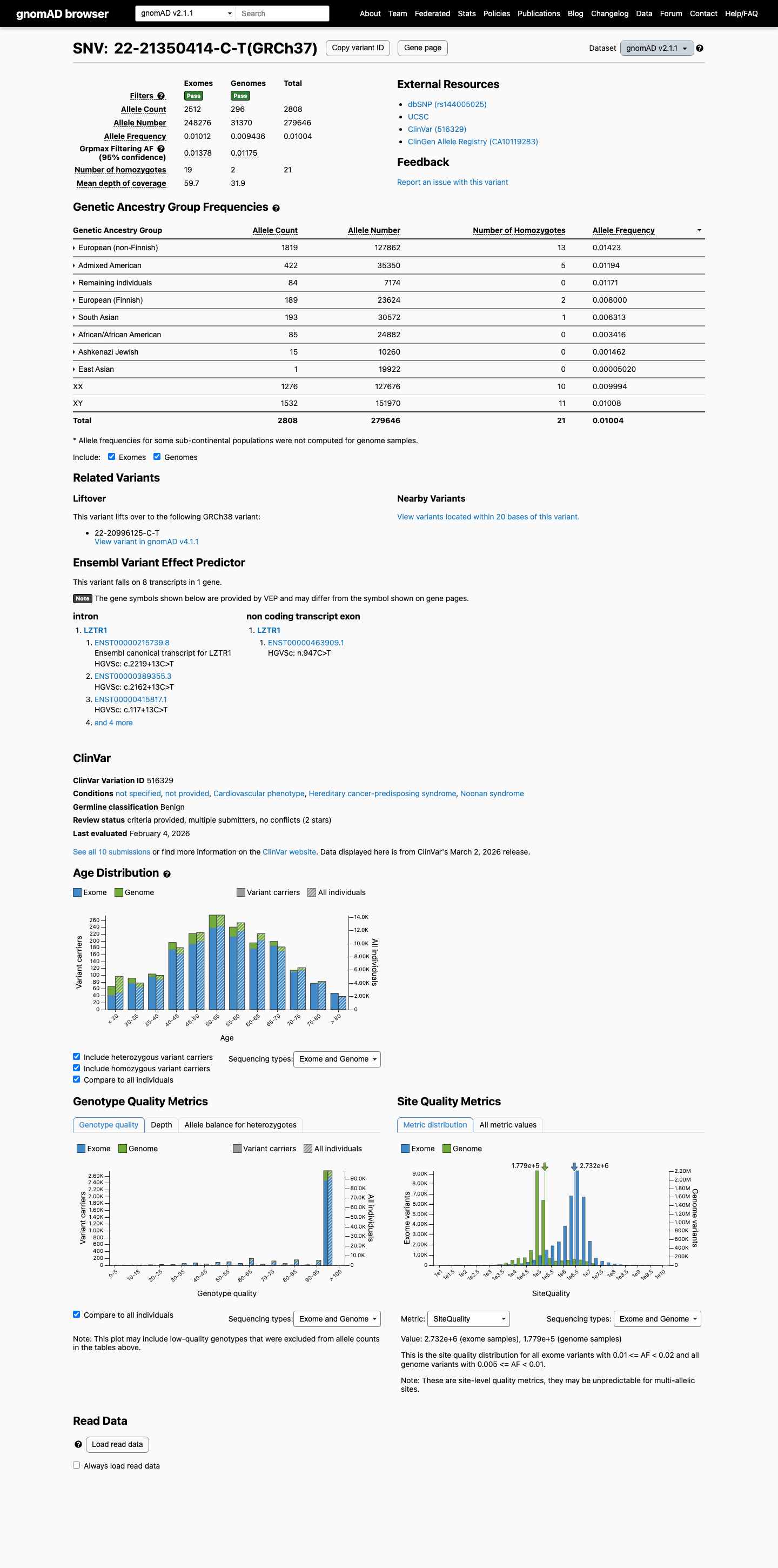
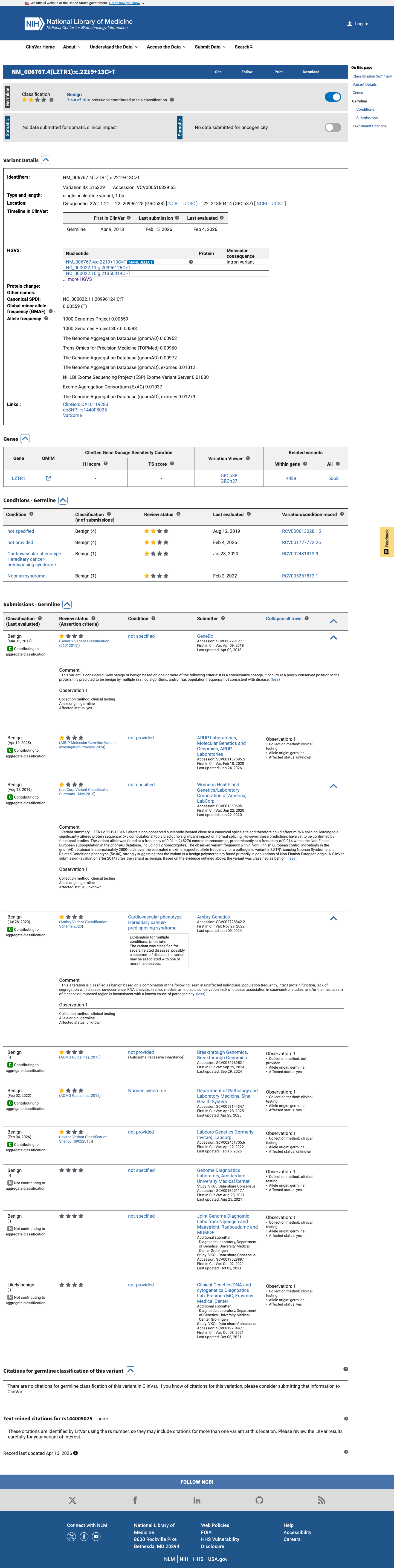
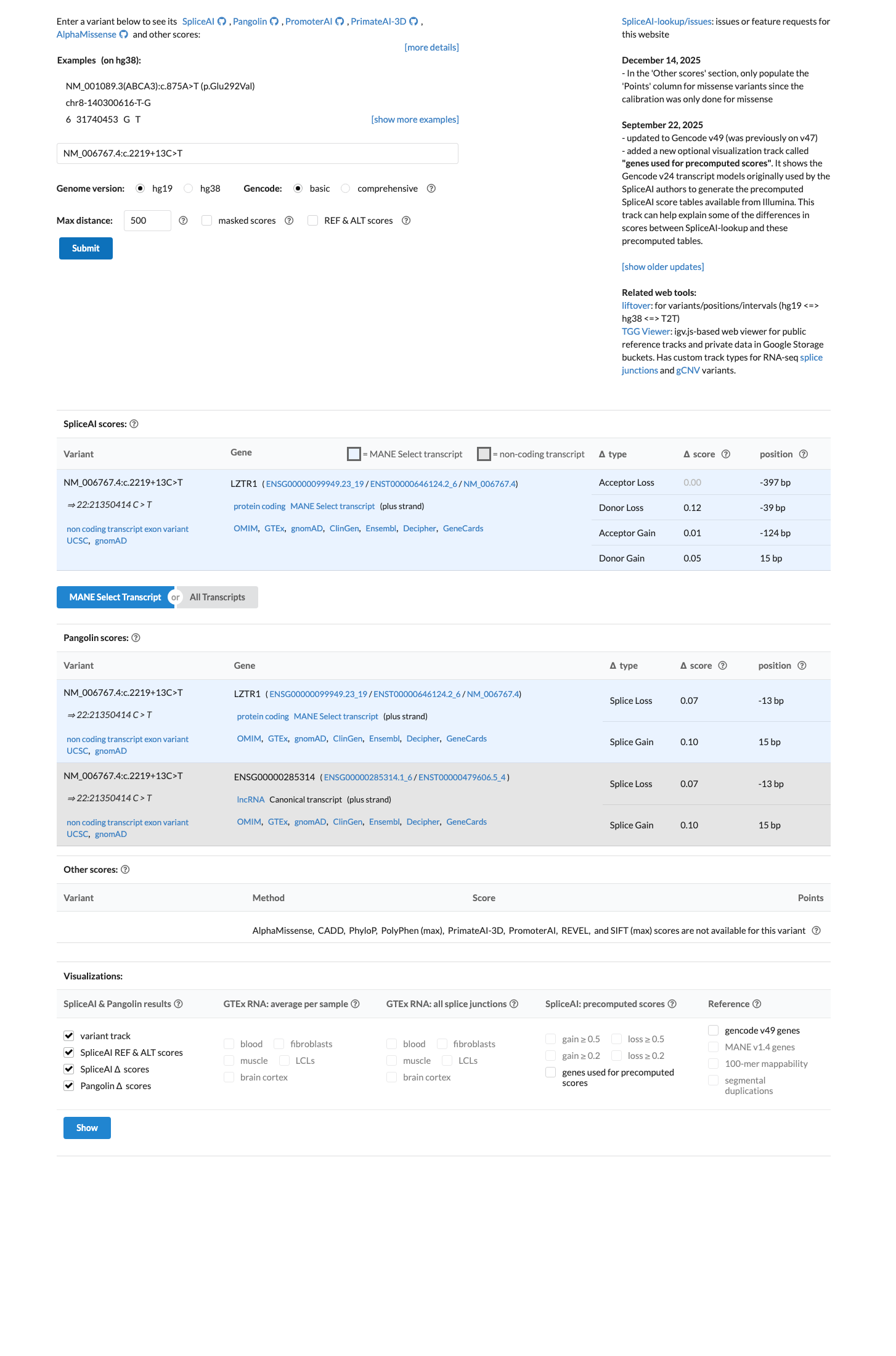
The LZTR1 c.2219+13C>T (p.?) variant has been reported in ClinVar predominantly as benign, with 8 benign and 1 likely benign clinical laboratory submissions.1 This variant is common in population databases, with gnomAD v2.1 grpmax filtering allele frequency 1.37781% and gnomAD v4.1 grpmax filtering allele frequency 1.43127%, both well above the LZTR1 benign thresholds for BS1 (0.025%) and BA1 (0.05%).2 SpliceAI predicts no significant splice impact for this variant, with a maximum delta score of 0.13, which is consistent with BP4 and BP7 rather than a deleterious splicing effect.3
BP4 + BS1 + BA1 + BP7
→
Benign