The CHEK2 c.715G>A (p.Glu239Lys, p.E239K) variant has been reported in ClinVar with predominantly uncertain significance submissions and one likely pathogenic submission.1 This variant is present in gnomAD at 0.00849% in v2.1 and 0.01091% in v4.1, with a highest observed subpopulation frequency of 0.03333%, which is below the 0.1% rarity threshold and does not reach BS1 or BA1 population thresholds.2 In a published yeast-based DNA-damage response assay, p.E239K had an intermediate score of 0.58 relative to 1.00 for wild type and 0.00 for a kinase-dead control, which is consistent with partial functional impairment but does not by itself establish a definitive functional criterion strength.3 Computational evidence is mixed: SpliceAI predicts no significant splice impact with a max delta score of 0.00, REVEL is 0.445, and BayesDel is 0.263648, so in silico evidence alone does not clearly support either a damaging or benign computational criterion.4
CHEK2
Final classification
VUS
CHEK2 c.715G>A · p.Glu239Lys
CHEK2
The CHEK2 c.715G>A (p.Glu239Lys, p.E239K) variant has been reported in ClinVar with predominantly uncertain significance submissions and one likely pathogenic submission.
Hereditary Breast, Ovarian and Pancreatic Cancer Specification v1.0.0 lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 moderate; combination = 1 moderate, which maps to VUS.
Classification rationale
PM2
VUS
CHEK2 c.715G>A
PM2
→
VUS
Gene diagram
· NM_007194.3 · variants mapped to exon structure
CHEK2
NM_007194.3
Fetching transcript structure from UCSC…
Applied criteria · 1 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency
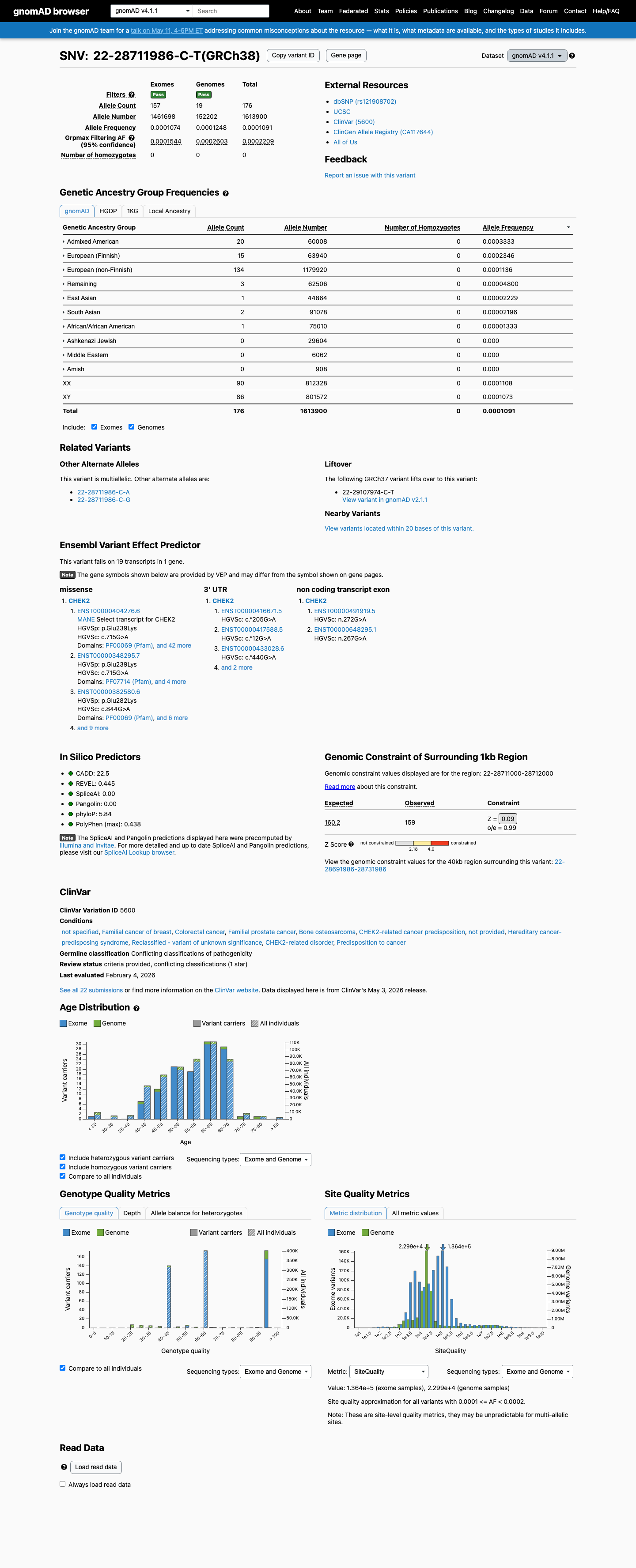
gnomAD v4.1
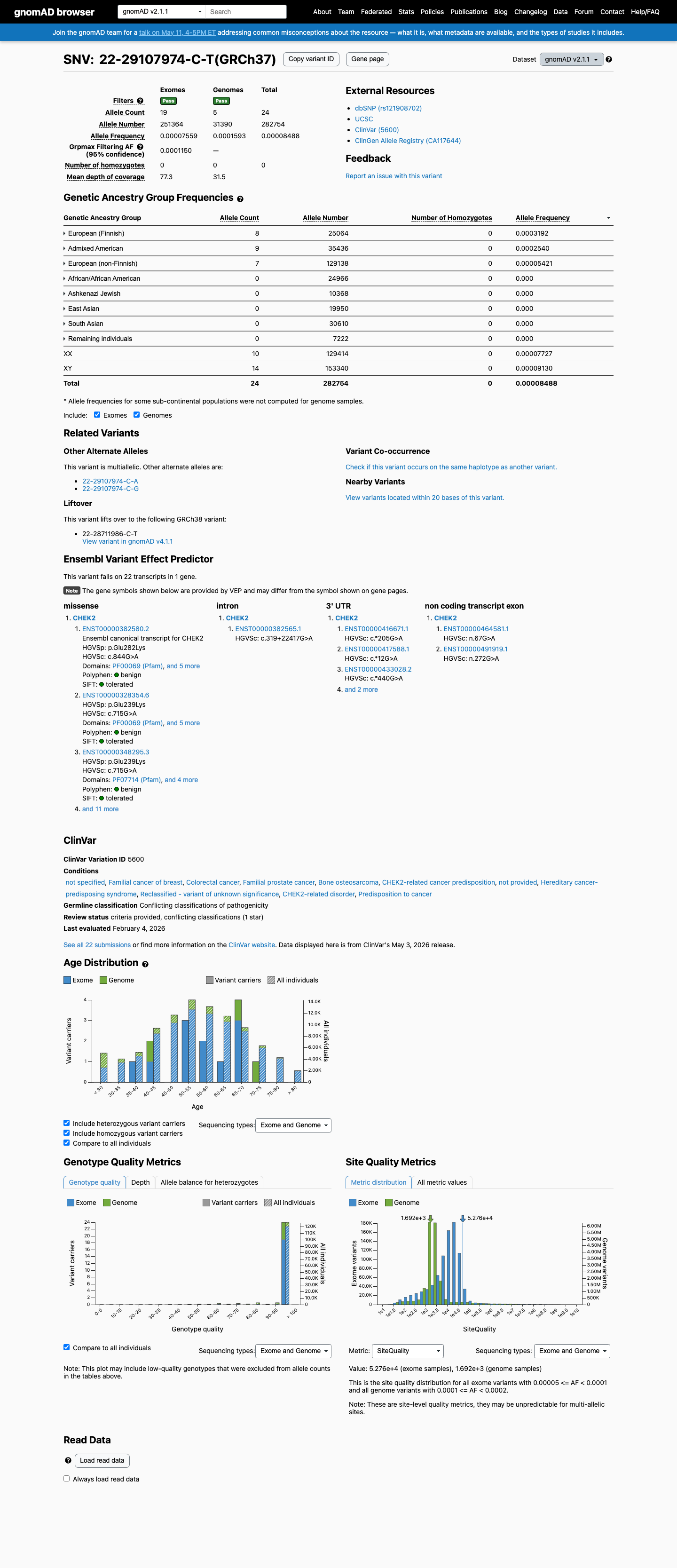
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000109053; MAF= 0.01091%, 176/1613900 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 0.000333289; MAF= 0.03333%, 20/60008 alleles, homozygotes = 0); grpmax FAF= 0.00022085.
v2.1
This variant is present in gnomAD v2.1 (AF= 8.48794e-05; MAF= 0.00849%, 24/282754 alleles, homozygotes = 0) and has highest observed frequency in the European (Finnish) population (AF= 0.000319183; MAF= 0.03192%, 8/25064 alleles, homozygotes = 0); grpmax FAF= 0.00011498.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.011%
· 176 / 1,613,900
0 hom · FAF 0.022%
0 hom · FAF 0.022%
Admixed American 20 / 60,008 |
0.033% |
European (Finnish) 15 / 63,940 |
0.023% |
European (non-Finnish) 134 / 1,179,920 |
0.011% |
Remaining individuals 3 / 62,506 |
0.0048% |
East Asian 1 / 44,864 |
0.0022% |
South Asian 2 / 91,078 |
0.0022% |
African/African American 1 / 75,010 |
0.0013% |
+ 3 not observed (Amish, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.0085%
· 24 / 282,754
0 hom · FAF 0.011%
0 hom · FAF 0.011%
European (Finnish) 8 / 25,064 |
0.032% |
Admixed American 9 / 35,436 |
0.025% |
European (non-Finnish) 7 / 129,138 |
0.0054% |
+ 5 not observed (African/African American, Ashkenazi Jewish, East Asian, Remaining individuals, South Asian)
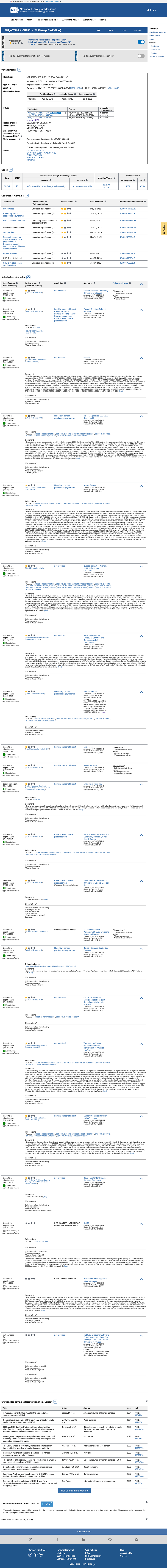
ClinVar
This variant has been reported in ClinVar as Uncertain significance (17 clinical laboratories) and as Likely pathogenic (1 clinical laboratory) and as uncertain significance (1 clinical laboratory). (ClinVarID = 5600)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.445. BayesDel score = 0.263648.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. CHEK2, an intracellular kinase involved in control of the cell cycle, is altered in various cancer types.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 10 PMIDs not cited in assessment
23298314 ↗
Proteome-wide analysis of amino acid variations that influence protein lysine acetylation.
CLINVAR
25525159 ↗
RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
26506619 ↗
Association of Germline CHEK2 Gene Variants with Risk and Prognosis of Non-Hodgkin Lymphoma.
CLINVAR
28166811 ↗
Pathogenic variant burden in the ExAC database: an empirical approach to evaluating population data for clinical variant interpretation.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR