NM_007194.4:c.1376-23G>C is a deep intronic variant in CHEK2 intron 12, located 23 bases upstream of exon 13. This variant is present at extremely low frequency in population databases: gnomAD v2.1 allele frequency = 0.0064% (15/233,494 alleles) and gnomAD v4.1 allele frequency = 0.0018% (27/1,501,478 alleles), with all observations restricted to the South Asian population and no homozygotes observed (PM2_Supporting).1 Computational splicing analysis with SpliceAI predicts no impact on splicing (max delta score = 0.00; all four delta scores for acceptor gain, acceptor loss, donor gain, and donor loss are 0.00), consistent with a benign computational profile (BP4_Supporting).2 The variant does not fall into any PVS1 null-variant category (not a nonsense, frameshift, or canonical splice variant), and SpliceAI confirms no predicted cryptic splice alteration.3 No functional studies, segregation data, de novo observations, case-control data, or ClinVar classifications are available for this variant. Applying the generic ACMG/AMP 2015 final combination rules (PMID:25741868), the evidence profile consists of one supporting pathogenic criterion (PM2) and one supporting benign criterion (BP4). With only one supporting criterion on each side, the evidence is indeterminate, resulting in a classification of Variant of Uncertain Significance (VUS).4
CHEK2
Final classification
VUS
CHEK2 c.1376-23G>C · p.?
CHEK2
NM_007194.4:c.1376-23G>C is a deep intronic variant in CHEK2 intron 12, located 23 bases upstream of exon 13.
Hereditary Breast, Ovarian and Pancreatic Cancer Specification v1.0.0 lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting benign; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP4
VUS
CHEK2 c.1376-23G>C
PM2 + BP4
→
VUS
3
pvs1_variant_assessmentspliceai ↗
4
generic_acmg_combination_rules
Gene diagram
· NM_007194.4 · variants mapped to exon structure
CHEK2
NM_007194.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 17 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is present at extremely low frequency in population databases: gnomAD v2.1 AF = 6.42e-5 (0.0064%, 15/233,494 alleles) and gnomAD v4.1 AF = 1.80e-5 (0.0018%, 27/1,501,478 alleles), both well below the PM2 threshold of 0.1%. The grpmax filtering allele frequency is 0.03% (v2.1) and 0.02% (v4.1), also below 0.1%. All observed alleles are restricted to the South Asian population. No homozygotes observed. Absent from gnomAD-Canada. Absent from ClinVar.
gnomAD v2.1: AF=6.42e-5 (15/233494)0 homozygotes
✓
BP4
supporting
Benign
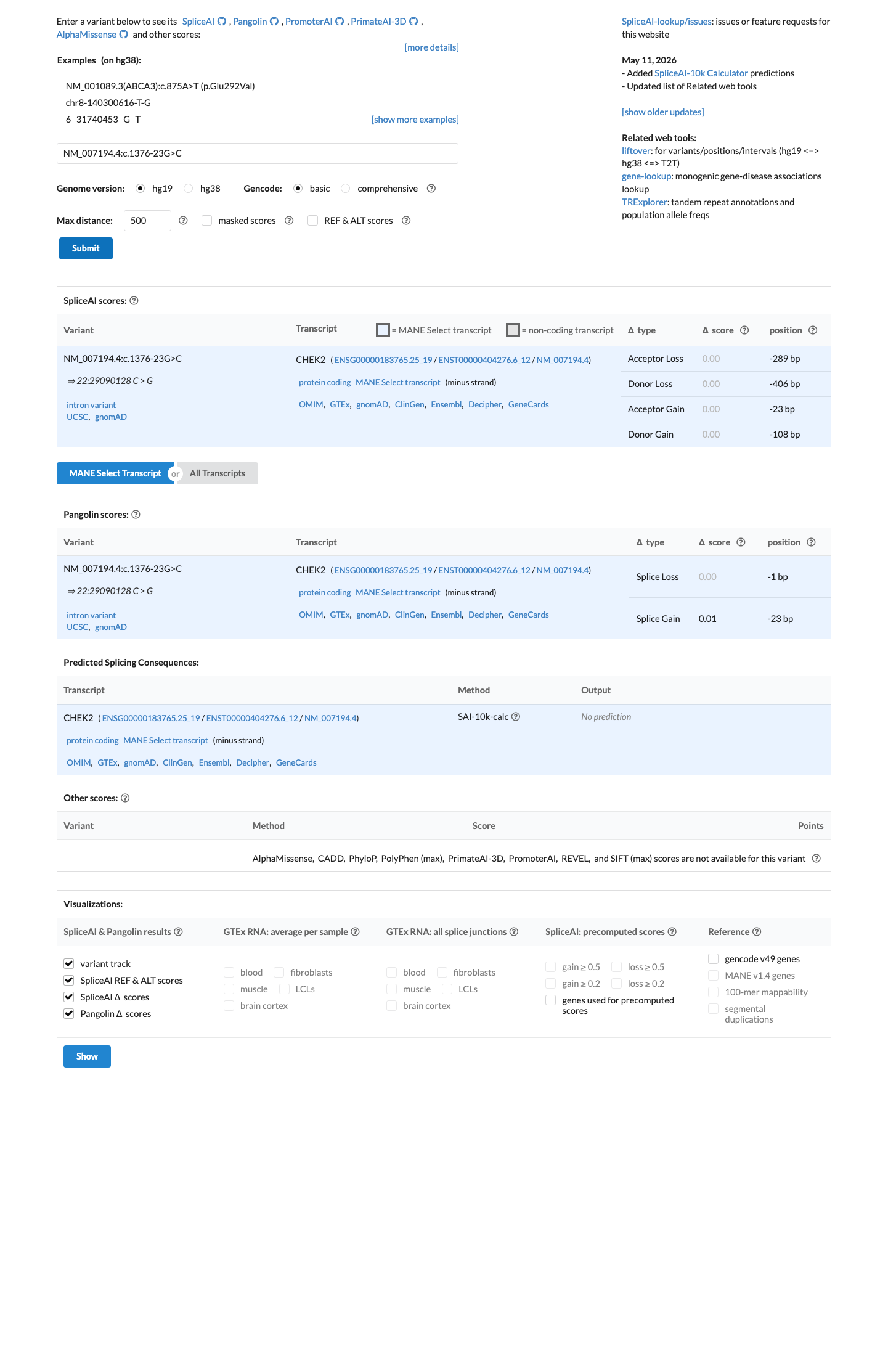
Multiple lines of computational evidence suggest no impact on gene product. SpliceAI predicts no splicing impact for this intronic variant (max delta score = 0.00; all acceptor and donor gain/loss scores are 0.00). REVEL and BayesDel are not applicable to intronic variants, and the absence of any predicted splice alteration is consistent with a benign computational profile.
SpliceAI max delta = 0.00all four delta scores (acceptor gainacceptor loss
Assessed · not applied
Pathogenic
PS2
No evidence of a de novo occurrence of this variant in a proband with CHEK2-related cancer phenotype.
PS3
No published functional studies (in vitro or in vivo) directly assessing the impact of NM_007194.4:c.1376-23G>C on CHEK2 splicing, expression, or kinase activity were identified.
PS4
No case-control study comparing the frequency of this variant in CHEK2-related cancer cohorts versus the general population is available.
PM1
This is a deep intronic variant (c.1376-23G>C) not located in a known mutational hot spot or critical functional domain.
PM6
No de novo observation of this variant has been reported without confirmation of paternity and maternity.
PP1
No cosegregation data are available for this variant in families with CHEK2-related cancer.
PP3
Multiple lines of computational evidence do not support a deleterious effect.
PP4
No patient phenotype or family history data specific to this variant are available for review.
PP5
No reputable source has recently reported this variant as pathogenic with unavailable supporting evidence.
Benign
BA1
The maximum allele frequency observed for this variant is 0.0495% (gnomAD v2.1 South Asian population), which is well below the BA1 threshold of 1% (or 5% by generic ACMG/AMP).
BS1
The maximum allele frequency of 0.0495% (gnomAD v2.1 SAS) is below the BS1 threshold of 0.3% for CHEK2-related cancer predisposition.
BS2
No evidence that this variant has been observed in a healthy adult individual in a manner inconsistent with pathogenic contribution (e.g., homozygous state, or in trans with a pathogenic variant for a recessive disorder).
BS3
No well-established functional studies demonstrate that this variant has no deleterious effect on CHEK2 splicing or protein function.
BS4
No family segregation data are available to demonstrate that this variant does not segregate with disease in affected family members.
BP2
No evidence that this variant has been observed in cis with a known pathogenic CHEK2 variant in a patient.
BP5
No evidence that this variant has been found in a case with an alternative molecular basis for disease.
BP6
No reputable source has recently reported this variant as benign with unavailable supporting evidence.
N/A · 9
PVS1 · PS1 · PM3 · PM4 · PM5 · PP2 · BP1 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
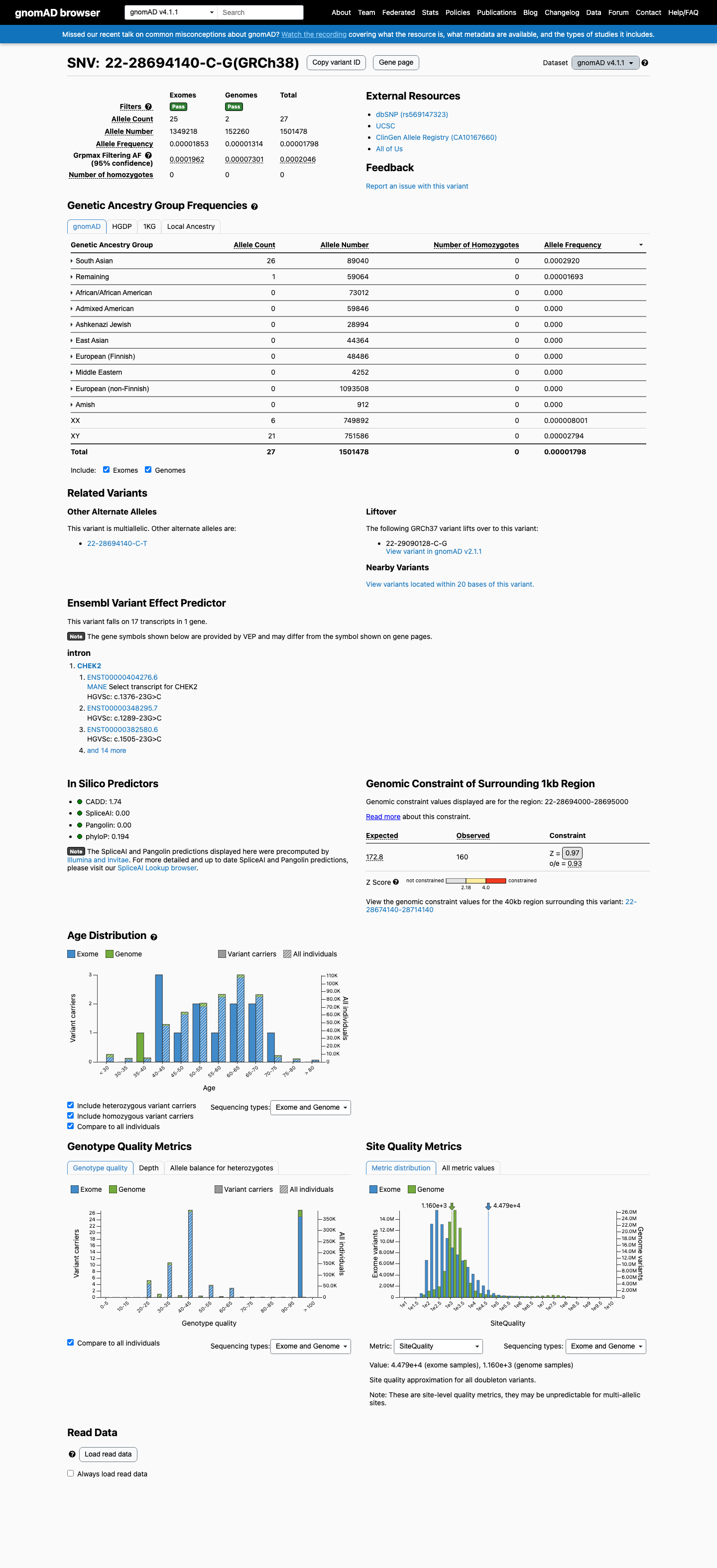
This variant is present in gnomAD v4.1 (AF= 1.79823e-05; MAF= 0.00180%, 27/1501478 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 0.000292004; MAF= 0.02920%, 26/89040 alleles, homozygotes = 0); grpmax FAF= 0.00020457.
v2.1
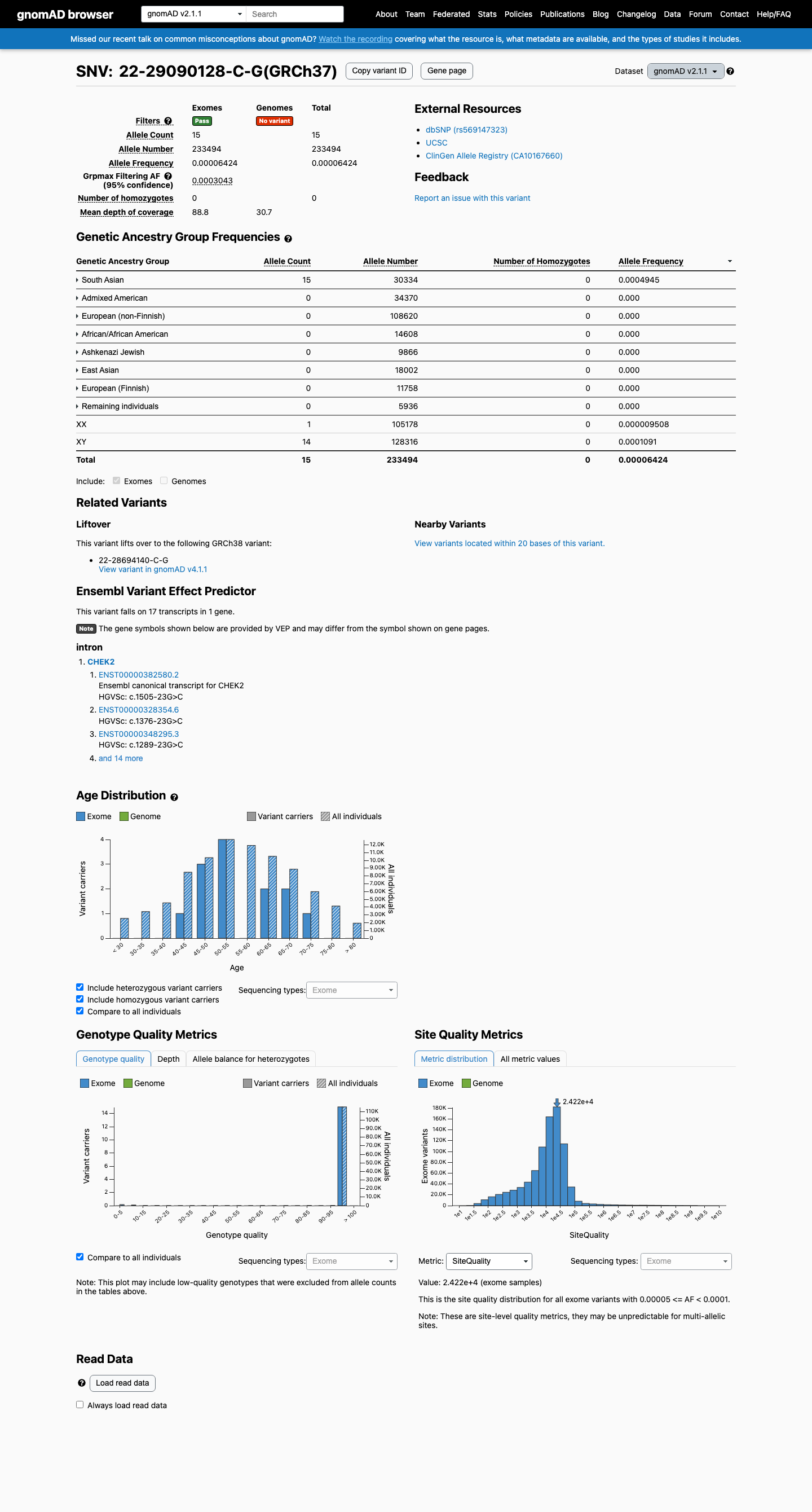
This variant is present in gnomAD v2.1 (AF= 6.42415e-05; MAF= 0.00642%, 15/233494 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 0.000494495; MAF= 0.04945%, 15/30334 alleles, homozygotes = 0); grpmax FAF= 0.00030425.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0018%
· 27 / 1,501,478
0 hom · FAF 0.02%
0 hom · FAF 0.02%
South Asian 26 / 89,040 |
0.029% |
Remaining individuals 1 / 59,064 |
0.0017% |
+ 8 not observed (Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish, African/African American, European (non-Finnish))
gnomAD v2.1
0.0064%
· 15 / 233,494
0 hom · FAF 0.03%
0 hom · FAF 0.03%
South Asian 15 / 30,334 |
0.049% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), Remaining individuals)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links