NM_007194.4:c.1427C>T (p.Thr476Met) is a missense variant in CHEK2 exon 13, located within the protein kinase domain. This variant is present in gnomAD v2.1 at an allele frequency of 0.0313% (83/265,178 alleles, 0 homozygotes) and in gnomAD v4.1 at 0.0404% (644/1,595,894 alleles) including one homozygous carrier in the Middle Eastern subpopulation (AF 0.407%).1 The variant is reported in ClinVar with conflicting classifications: Uncertain significance (29 clinical laboratories), Likely pathogenic (22 laboratories), and single submissions of Pathogenic, Likely benign, Likely risk allele, and Established risk allele (ClinVar Variation ID: 128060).2 The Middle Eastern subpopulation allele frequency of 0.407% in gnomAD v4.1 exceeds the BS1 threshold of 0.3%, which is higher than expected for a pathogenic CHEK2 variant (BS1_Supporting).3 Multiple in silico tools predict a benign effect: BayesDel score 0.096 is in the benign range and SpliceAI max delta 0.03 predicts no splicing alteration. REVEL score 0.445 is borderline (BP4_Supporting).4 The variant was identified as a novel missense substitution in a Bulgarian breast cancer cohort (PMID:22862163, Angelova et al. 2012, n=145 patients), but no case-control statistics or functional characterization were provided. No well-established functional assay data, segregation studies, or case-control analyses are available to confirm or refute pathogenicity for this specific variant. The presence of a homozygous carrier in gnomAD v4.1 is notable for an autosomal dominant cancer predisposition gene, though formal BS2 criteria are not met because CHEK2-related cancer risk has incomplete penetrance and adult onset, and the phenotype of the homozygous individual is unknown.5 Overall, the available evidence includes one supporting benign criterion (BS1) and one supporting benign criterion (BP4), with no pathogenic criteria met. The evidence favors a benign interpretation but remains insufficient for a definitive classification.
CHEK2
Final classification
Likely Benign
CHEK2 c.1427C>T · p.Thr476Met
CHEK2
NM_007194.4:c.1427C>T (p.Thr476Met) is a missense variant in CHEK2 exon 13, located within the protein kinase domain.
Hereditary Breast, Ovarian and Pancreatic Cancer Specification v1.0.0 lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS1 supporting benign, BP4 supporting benign; combination = 2 supporting benign, which maps to Likely Benign.
Classification rationale
BS1BP4
Likely Benign
CHEK2 c.1427C>T
BS1 + BP4
→
Likely Benign
Gene diagram
· NM_007194.4 · variants mapped to exon structure
CHEK2
NM_007194.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 19 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
supporting
Benign
The gnomAD v4.1 Middle Eastern subpopulation allele frequency of 0.407% exceeds the BS1 threshold of 0.3% for a dominant disorder. This frequency is higher than expected for a pathogenic CHEK2 variant with moderate penetrance, particularly given the presence of a homozygous carrier in the same subpopulation.
gnomAD v4.1 Middle Eastern subpopulation AF 0.407% (18/4424 alleles) exceeds BS1 threshold of 0.3%one homozygous carrier observed.
✓
BP4
supporting
Benign
Multiple lines of computational evidence suggest a benign effect. BayesDel score 0.096 falls within the benign range. SpliceAI max delta score 0.03 predicts no splicing alteration. REVEL score 0.445 is borderline and does not independently predict pathogenicity. Two of three computational tools support a benign interpretation.
BayesDel 0.096 (benignbelow 0.13 threshold)SpliceAI max delta 0.03 (no splicing impact)
Assessed · not applied
Pathogenic
PS1
No known pathogenic variant at the same amino acid position (Thr476) with a different nucleotide change has been identified.
PS2
No documented de novo occurrence of NM_007194.4:c.1427C>T has been identified in the literature or databases.
PS3
No well-established functional assay data demonstrating a damaging effect for p.Thr476Met has been confirmed.
PS4
No case-control study demonstrating statistically significant enrichment of NM_007194.4:c.1427C>T in affected individuals versus controls has been identified.
PM1
Although p.Thr476Met is located within the CHEK2 protein kinase domain (residues ~229-507), a critical functional domain, the presence of a homozygous carrier in gnomAD v4.1 (Middle Eastern population) and a subpopulation allele frequency of 0.407% indicate that this residue tolerates variation in the general population.
PM2
While gnomAD v2.1 overall allele frequency (0.0313%) is below the generic PM2 threshold of 0.1%, gnomAD v4.1 data reveal a higher frequency (0.0404% overall; 0.407% in the Middle Eastern subpopulation) including one homozygous carrier.
PM6
No assumed de novo occurrence (unconfirmed parentage) has been reported for this variant.
PP1
No cosegregation data have been published for NM_007194.4:c.1427C>T with disease in multiple affected family members.
PP2
PP2 requires a low rate of benign missense variation in the gene and that missense variants are a common mechanism of disease.
PP3
Multiple computational tools do not support a deleterious effect.
PP4
No patient-specific phenotype or family history data were provided for this case.
PP5
No reputable source has definitively classified this variant as pathogenic.
Benign
BA1
The highest observed population allele frequency is 0.407% in the gnomAD v4.1 Middle Eastern subpopulation, which does not exceed the 1% threshold required for BA1.
BS2
Although one homozygous carrier is present in gnomAD v4.1 (Middle Eastern population), BS2 requires observation in a healthy adult with a disorder expected to have full penetrance at an early age.
BS3
No well-established functional assay demonstrating a benign effect for p.Thr476Met has been identified.
BS4
No segregation data demonstrating lack of cosegregation with disease have been published for this variant.
BP2
No observation of this variant in trans with a known pathogenic CHEK2 variant (in a recessive model) or in cis with a pathogenic variant (in a dominant model) has been reported.
BP5
No evidence is available indicating that this variant was found in a case with an alternate molecular basis for disease.
BP6
No reputable source has definitively classified this variant as benign.
N/A · 4
PVS1 · PM5 · BP1 · BP7
Research & evidence
Population frequency · supports benign
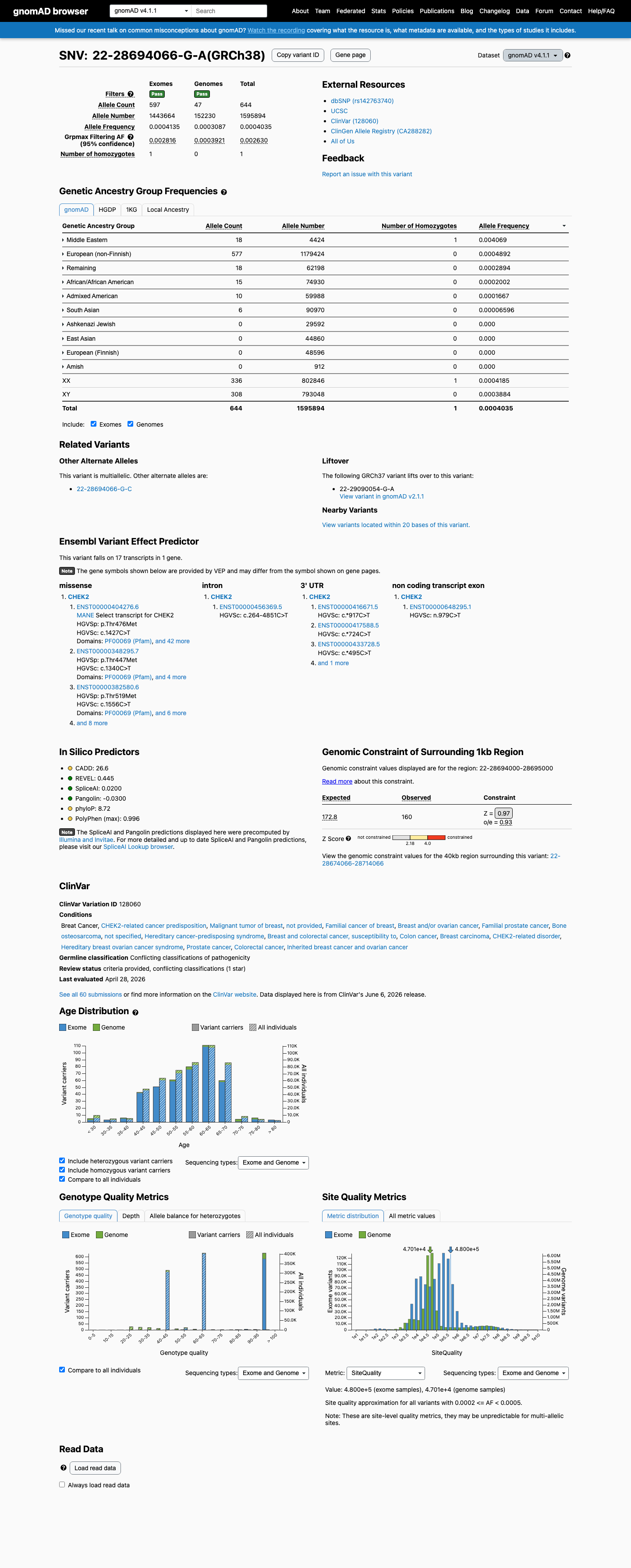
gnomAD v4.1
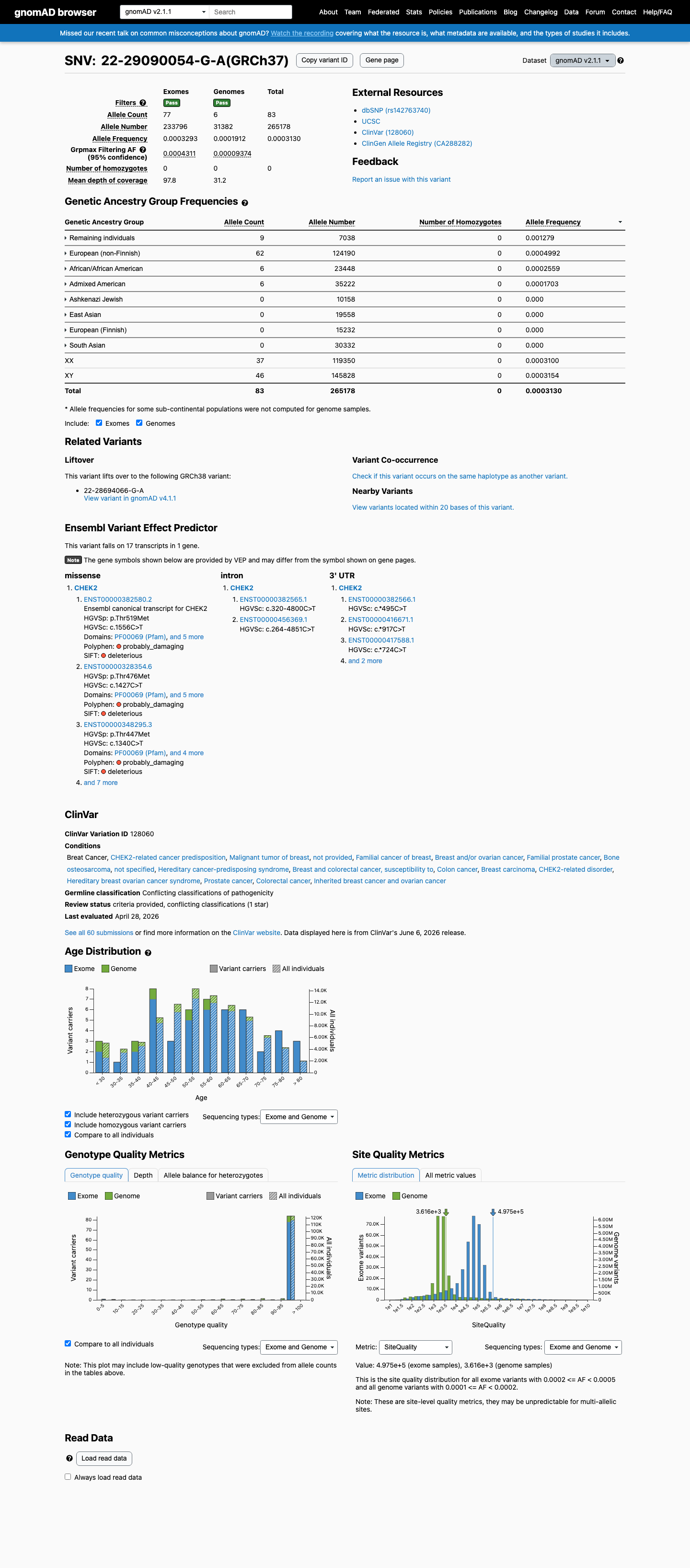
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000403536; MAF= 0.04035%, 644/1595894 alleles, homozygotes = 1) and has highest observed frequency in the Middle Eastern population (AF= 0.00406872; MAF= 0.40687%, 18/4424 alleles, homozygotes = 1); grpmax FAF= 0.00262965.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.000312997; MAF= 0.03130%, 83/265178 alleles, homozygotes = 0) and has highest observed frequency in the Remaining individuals population (AF= 0.00127877; MAF= 0.12788%, 9/7038 alleles, homozygotes = 0); grpmax FAF= 0.0004311.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.04%
· 644 / 1,595,894
1 hom · FAF 0.26%
1 hom · FAF 0.26%
Middle Eastern 18 / 4,424 |
0.41% 1 hom |
European (non-Finnish) 577 / 1,179,424 |
0.049% |
Remaining individuals 18 / 62,198 |
0.029% |
African/African American 15 / 74,930 |
0.02% |
Admixed American 10 / 59,988 |
0.017% |
South Asian 6 / 90,970 |
0.0066% |
+ 4 not observed (European (Finnish), Amish, East Asian, Ashkenazi Jewish)
gnomAD v2.1
0.031%
· 83 / 265,178
0 hom · FAF 0.043%
0 hom · FAF 0.043%
Remaining individuals 9 / 7,038 |
0.13% |
European (non-Finnish) 62 / 124,190 |
0.05% |
African/African American 6 / 23,448 |
0.026% |
Admixed American 6 / 35,222 |
0.017% |
+ 4 not observed (Ashkenazi Jewish, East Asian, European (Finnish), South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

ClinVar
This variant has been reported in ClinVar as Uncertain significance (29 clinical laboratories) and as Likely pathogenic (22 clinical laboratories) and as Likely risk allele (1 clinical laboratory) and as Established risk allele (1 clinical laboratory) and as Pathogenic (1 clinical laboratory) and as Likely Pathogenic (1 clinical laboratory) and as Likely benign (1 clinical laboratory). (ClinVarID = 128060)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.03). REVEL score = 0.445. BayesDel score = 0.0956883.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. CHEK2, an intracellular kinase involved in control of the cell cycle, is altered in various cancer types.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 10 PMIDs not cited in assessment
23552953 ↗
Dissecting the genotype in syndromic intellectual disability using whole exome sequencing in addition to genome-wide copy number analysis.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
26845104 ↗
Improving performance of multigene panels for genomic analysis of cancer predisposition.
CLINVAR
27621404 ↗
Conflicting Interpretation of Genetic Variants and Cancer Risk by Commercial Laboratories as Assessed by the Prospective Registry of Multiplex Testing.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR