NM_007194.4:c.176C>A (p.Thr59Lys) is a missense variant in exon 2 of CHEK2, located in the N-terminal SQ/TQ regulatory domain. This variant has been observed in gnomAD at extremely low frequency: v2.1 AF=0.00159% (4/251,456 alleles) and v4.1 AF=0.00384% (62/1,613,896 alleles), meeting PM2 at supporting strength.1 T59K was directly tested in a yeast-based DNA damage response complementation assay and demonstrated intermediate functional impairment (score 0.51, normalized to wild-type=1.00 and kinase-dead control=0.00), consistent with partial loss of CHEK2-mediated DNA damage response. This meets PS3 at moderate strength (single publication, direct variant testing in a heterologous functional system).2 CHEK2 T59K was originally reported in four Icelandic breast cancer patients and additional individuals with colon, ovarian, and gastric cancers, although at frequencies not significantly different from controls.3 In silico predictions are conflicting and do not provide consensus: REVEL score is borderline (0.513), PolyPhen-2 predicts benign (0.30), BayesDel is below damaging threshold (0.410), and SIFT provides no prediction for this residue. SpliceAI predicts no splicing impact (max delta 0.00).4 The ClinGen Hereditary Breast, Ovarian and Pancreatic Cancer Expert Panel CHEK2 specification (VCEP v1.0) provides no per-criterion rules; generic ACMG/AMP 2015 criteria were applied.5 No additional pathogenic or likely pathogenic criteria were met. The variant is classified as a Variant of Uncertain Significance (VUS) based on one moderate criterion (PS3) and one supporting criterion (PM2).
CHEK2
Final classification
VUS
CHEK2 c.176C>A · p.Thr59Lys
CHEK2
NM_007194.4:c.176C>A (p.Thr59Lys) is a missense variant in exon 2 of CHEK2, located in the N-terminal SQ/TQ regulatory domain.
ClinGen Hereditary Breast, Ovarian and Pancreatic Cancer Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for CHEK2 Version 1.0 v1.0 lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PS3 moderate, PM2 supporting; combination = 1 moderate + 1 supporting, which maps to VUS.
Classification rationale
PS3PM2
VUS
CHEK2 c.176C>A
PS3 + PM2
→
VUS
4
revelbayesdelspliceai ↗PMID:22419737 ↗
5
cspec ↗
Gene diagram
· NM_007194.4 · variants mapped to exon structure
CHEK2
NM_007194.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 20 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PS3
moderate
Pathogenic
CHEK2 T59K was directly tested in a yeast-based DNA damage response complementation assay (Roeb et al. 2012, PMID:22419737) and demonstrated intermediate functional impairment (score 0.51 on a scale normalized to 1.00 wild-type and 0.00 kinase-dead control). The variant was significantly impaired relative to wild-type but retained partial function. Single publication with direct variant testing in a heterologous system qualifies for moderate-strength PS3.
Direct variant testing in yeast rad53 complementation assay: T59K showed intermediate DNA damage response (score 0.51)significantly different from both wild-type (1.00) and kinase-dead negative control (0.00).
✓
PM2
supporting
review
Pathogenic
NM_007194.4:c.176C>A is present in gnomAD at extremely low frequency: v2.1 AF=0.00159% (4/251,456 alleles, 0 homozygotes) and v4.1 AF=0.00384% (62/1,613,896 alleles, 0 homozygotes). The grpmax filtering allele frequency is 1.121e-05 in v2.1 and 4.019e-05 in v4.1. All values are well below the 0.1% PM2 threshold. No homozygotes observed.
gnomAD v2.1: AF=0.00159%4/251456 alleles
Assessed · not applied
Pathogenic
PS1
No evidence that a different nucleotide change at the same position produces the same amino acid change (p.Thr59Lys) with a known pathogenic classification.
PS2
No confirmed de novo observation of NM_007194.4:c.176C>A has been reported in the literature or ClinVar submissions.
PS4
No statistically significant case-control enrichment has been demonstrated for NM_007194.4:c.176C>A.
PM1
T59K is located in the N-terminal SQ/TQ domain (residues ~19-69) of CHEK2, an unstructured regulatory region containing ATM/ATR phosphorylation motifs.
PM6
No confirmed de novo observation of NM_007194.4:c.176C>A has been reported.
PP1
Familial segregation data reported by Ingvarsson et al.
PP2
PP2 is applicable to genes where missense variants are a predominant disease mechanism and the gene has a low rate of benign missense variation.
PP3
In silico predictions are conflicting and do not reach consensus.
PP4
No evidence that the patient's phenotype or family history is highly specific for CHEK2-related cancer predisposition relative to other hereditary cancer syndromes.
PP5
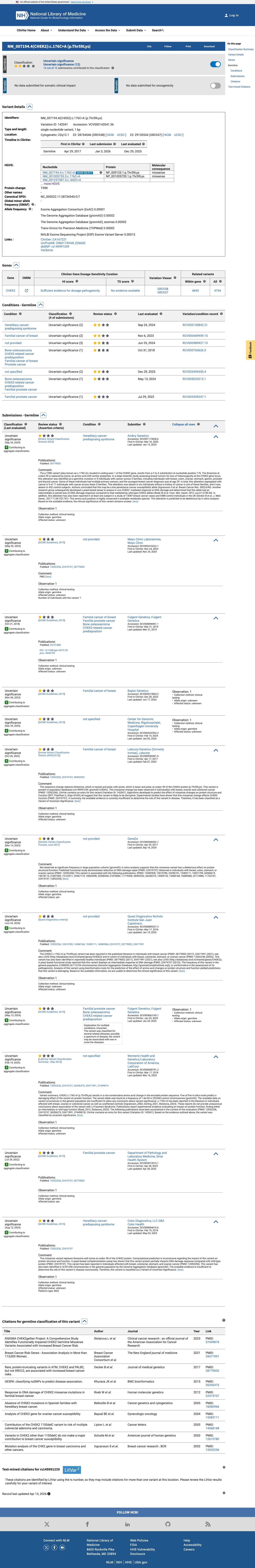
ClinVar variation ID 142041 is classified as Uncertain Significance by 12 clinical laboratories with review status 'criteria provided, single submitter.' There is no expert panel (3-star) classification.
Benign
BA1
The maximum allele frequency in gnomAD is 0.00508% (NFE, v4.1), well below the 1% BA1 threshold.
BS1
The maximum allele frequency in gnomAD is 0.00508% (NFE, v4.1), well below the 0.3% BS1 threshold.
BS2
No evidence that T59K has been observed in healthy adults at an age where full penetrance of CHEK2-related cancer predisposition would be expected, with a frequency sufficient to rule out pathogenicity.
BS3
The only published functional assay for T59K (PMID:22419737) demonstrated intermediate functional impairment (score 0.51), not wild-type-like function.
BS4
No evidence of lack of segregation with disease in affected families.
BP1
BP1 applies when a missense variant is found in a gene for which only truncating variants are known to cause disease.
BP2
No evidence of NM_007194.4:c.176C>A occurring in trans with a known pathogenic CHEK2 variant has been reported.
BP4
While some in silico tools predict a benign effect (PolyPhen-2: 0.30 benign; SpliceAI: 0.00), other predictors are conflicting or borderline (REVEL: 0.513; Align-GVGD: C65 highest risk).
BP5
No observation of NM_007194.4:c.176C>A occurring in trans with a known pathogenic CHEK2 variant has been reported.
BP6
No reputable source classifies NM_007194.4:c.176C>A as benign or likely benign.
N/A · 4
PVS1 · PM5 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
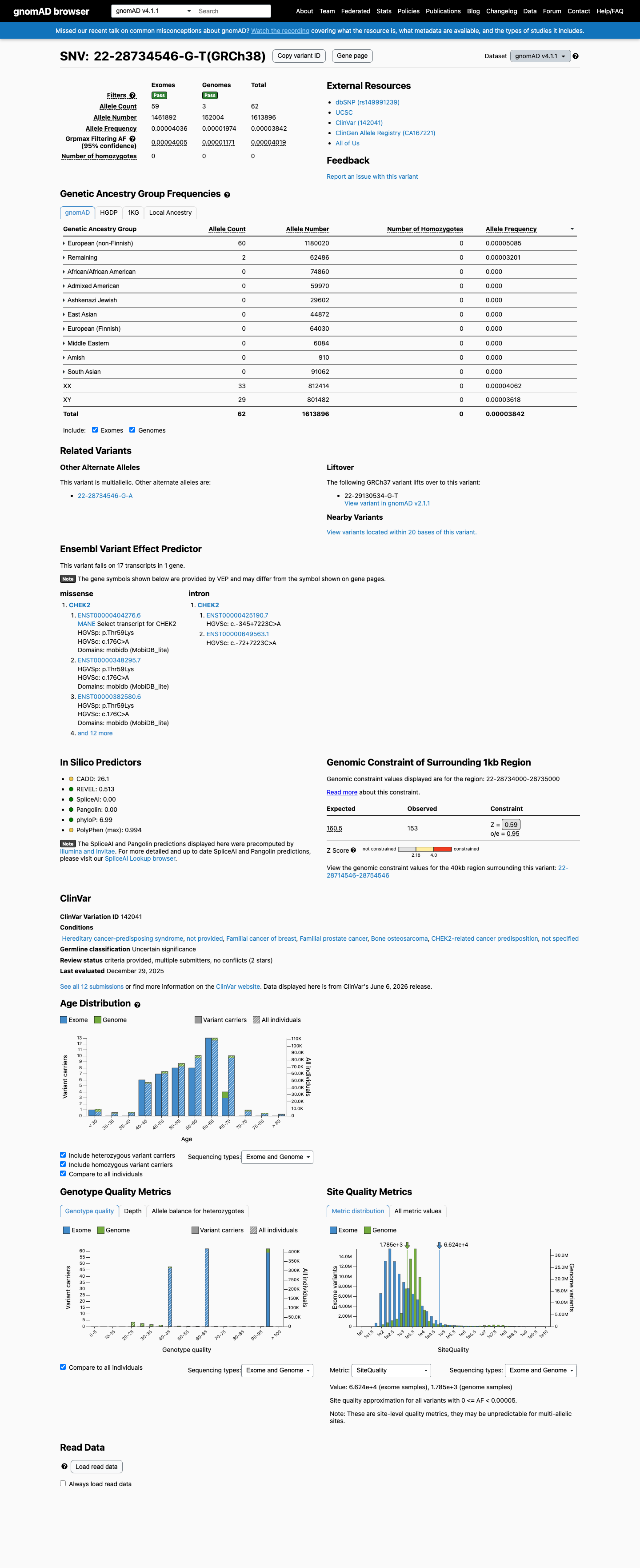
This variant is present in gnomAD v4.1 (AF= 3.84164e-05; MAF= 0.00384%, 62/1613896 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 5.08466e-05; MAF= 0.00508%, 60/1180020 alleles, homozygotes = 0); grpmax FAF= 4.019e-05.
v2.1
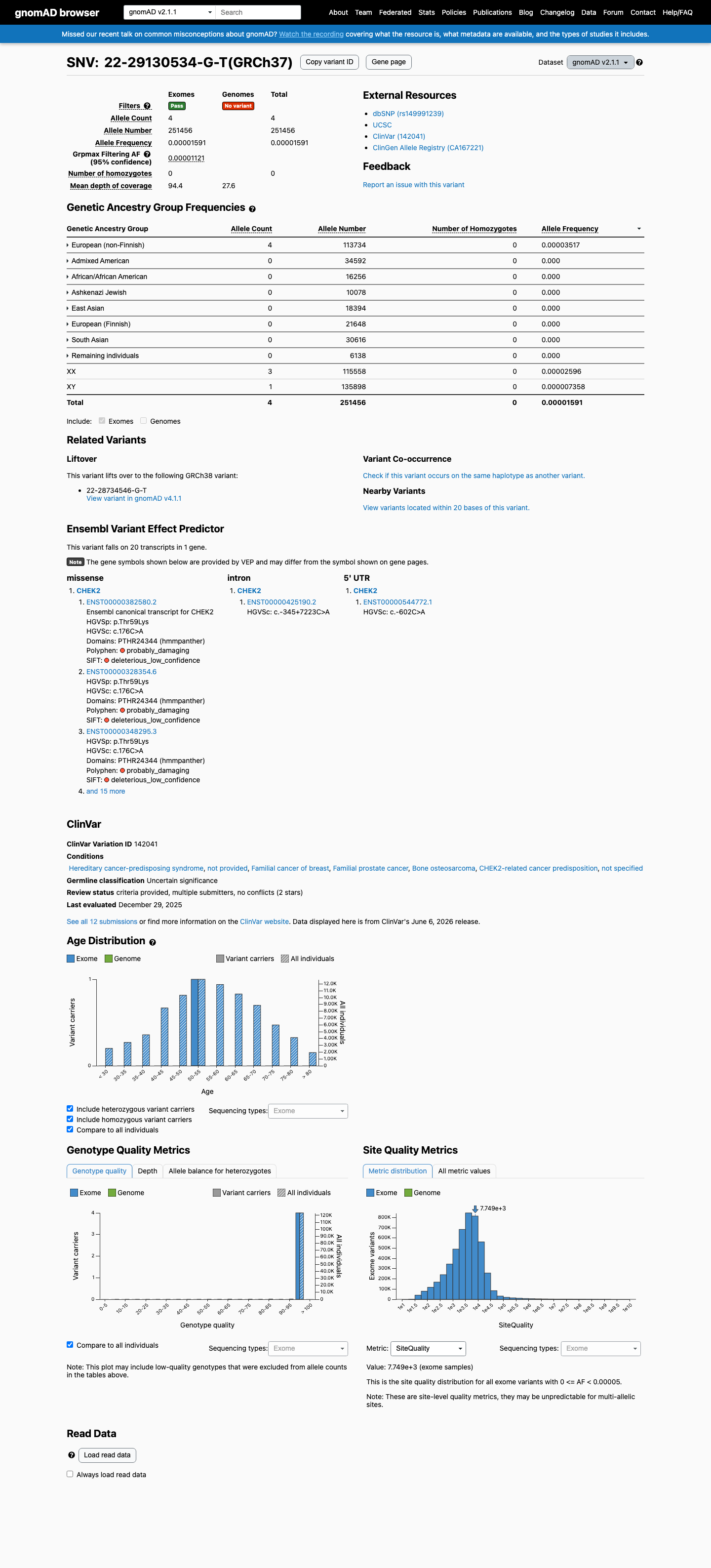
This variant is present in gnomAD v2.1 (AF= 1.59074e-05; MAF= 0.00159%, 4/251456 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 3.51698e-05; MAF= 0.00352%, 4/113734 alleles, homozygotes = 0); grpmax FAF= 1.121e-05.
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0038%
· 62 / 1,613,896
0 hom · FAF 0.004%
0 hom · FAF 0.004%
European (non-Finnish) 60 / 1,180,020 |
0.0051% |
Remaining individuals 2 / 62,486 |
0.0032% |
+ 8 not observed (Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0016%
· 4 / 251,456
0 hom · FAF 0.0011%
0 hom · FAF 0.0011%
European (non-Finnish) 4 / 113,734 |
0.0035% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Uncertain significance (12 clinical laboratories). (ClinVarID = 142041)
In silico
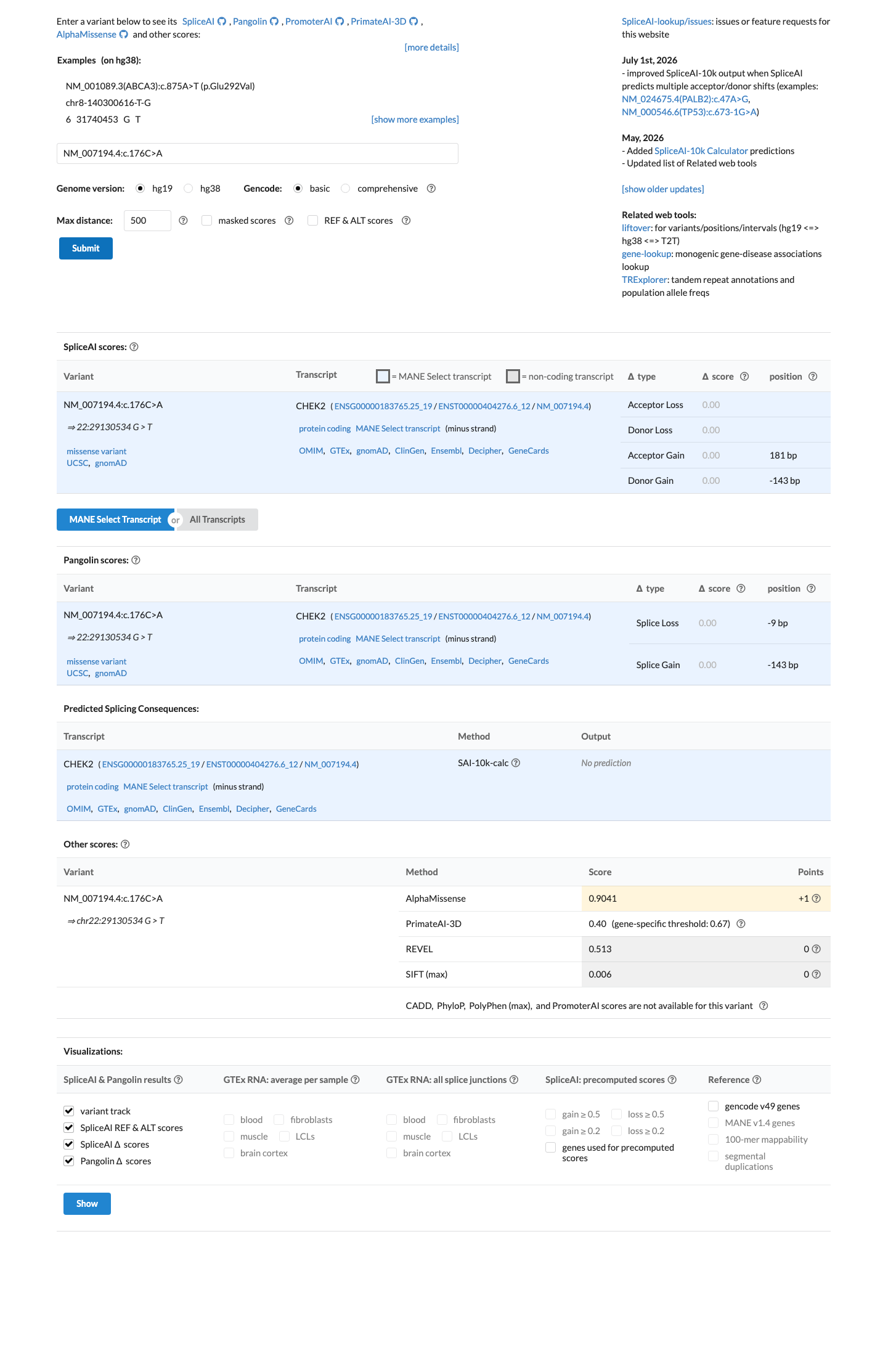
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.513. BayesDel score = 0.410012.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. CHEK2, an intracellular kinase involved in control of the cell cycle, is altered in various cancer types.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 8 further PMIDs triaged but not cited — see Sources & References.
Response to DNA damage of CHEK2 missense mutations in familial breast cancer.
Searched
c.176C>AT59KThr59Lys176C.A
Found
Roeb et al. developed a yeast-based assay to assess CHEK2-mediated DNA damage response for 25 germline missense variants identified in familial breast cancer patients. T59K (c.176C>A) was directly tested and demonstrated intermediate functional impairment with a quantitative score of 0.51 (normalized to wild-type=1.00 and kinase-dead D347A=0.00). The variant was classified as 'intermediate' — significantly impaired relative to wild-type but retaining partial function above the negative control. In silico predictions were inconclusive: PolyPhen-2 predicted benign (0.30), SIFT had no prediction, Align-GVGD assigned C65 (highest risk class), with an overall in silico consensus of 'no consensus.'
Variant
✓ Names this variant — characterised directly
Applied to
→PS3 supports · met
Why
Variant-specific functional data confirmed intermediate partial loss of function; supports PS3 at moderate strength. The SQ/TQ domain location does not independently meet PM1.
c.176C>A, T59K, SQ/TQ domain, PolyPhen2: 0.30 Benign, SIFT: no prediction, in silico consensus: nc, Present results: 0.51
Location Table 1 (rows for position 22:29,130,534 G>T, c.176C>A, T59K); Results; Discussion · Context Yeast (S. cerevisiae) rad53/sml1 deletion complementation assay; DNA damage induced by MMS; growth measured as functional readout; 2-4 independent experiments with 6 replicates each · full text
Sources & reference links
9Sources
Triaged references · 8 PMIDs not cited in assessment
12610780 ↗
Variants in CHEK2 other than 1100delC do not make a major contribution to breast cancer susceptibility.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
28779002 ↗
Rare, protein-truncating variants in ATM, CHEK2 and PALB2, but not XRCC2, are associated with increased breast cancer risks.
CLINVAR
37449874 ↗
ENIGMA CHEK2gether Project: A Comprehensive Study Identifies Functionally Impaired CHEK2 Germline Missense Variants Associated with Increased Breast Cancer Risk.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR