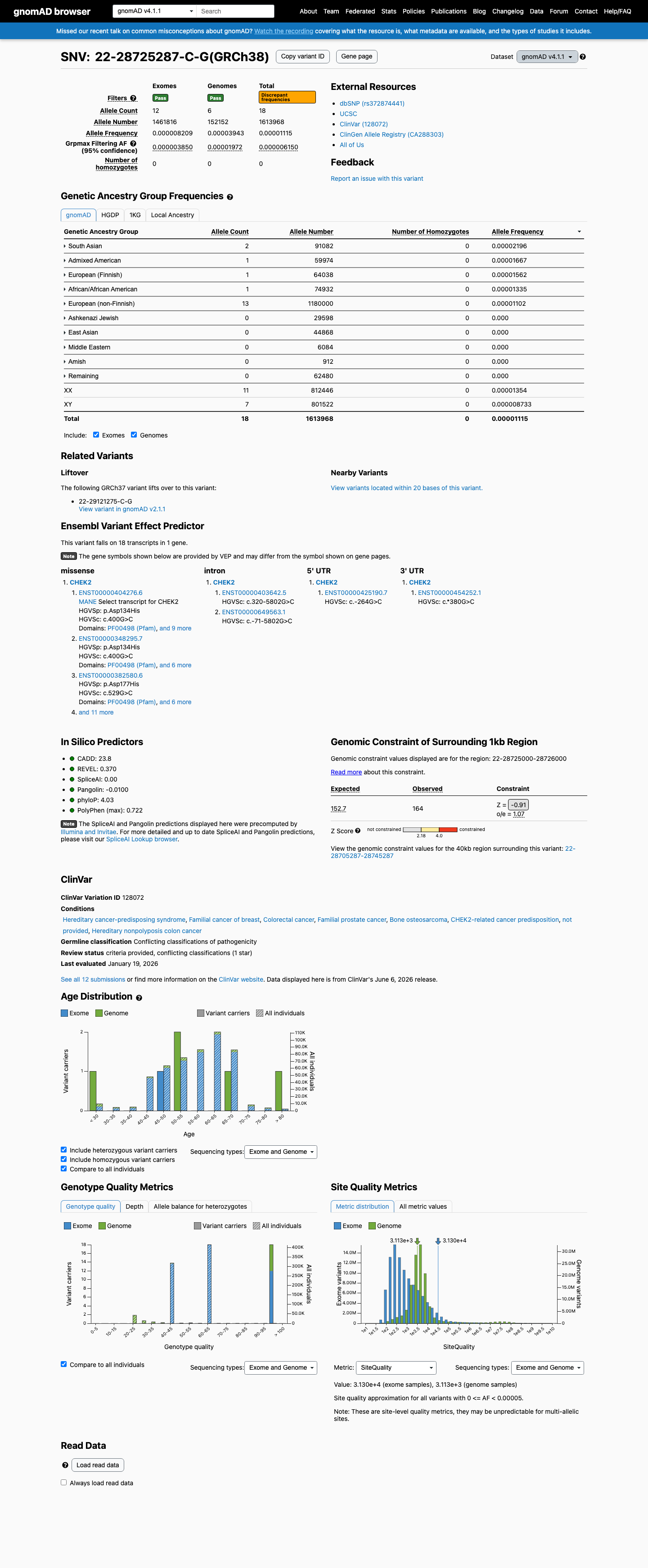
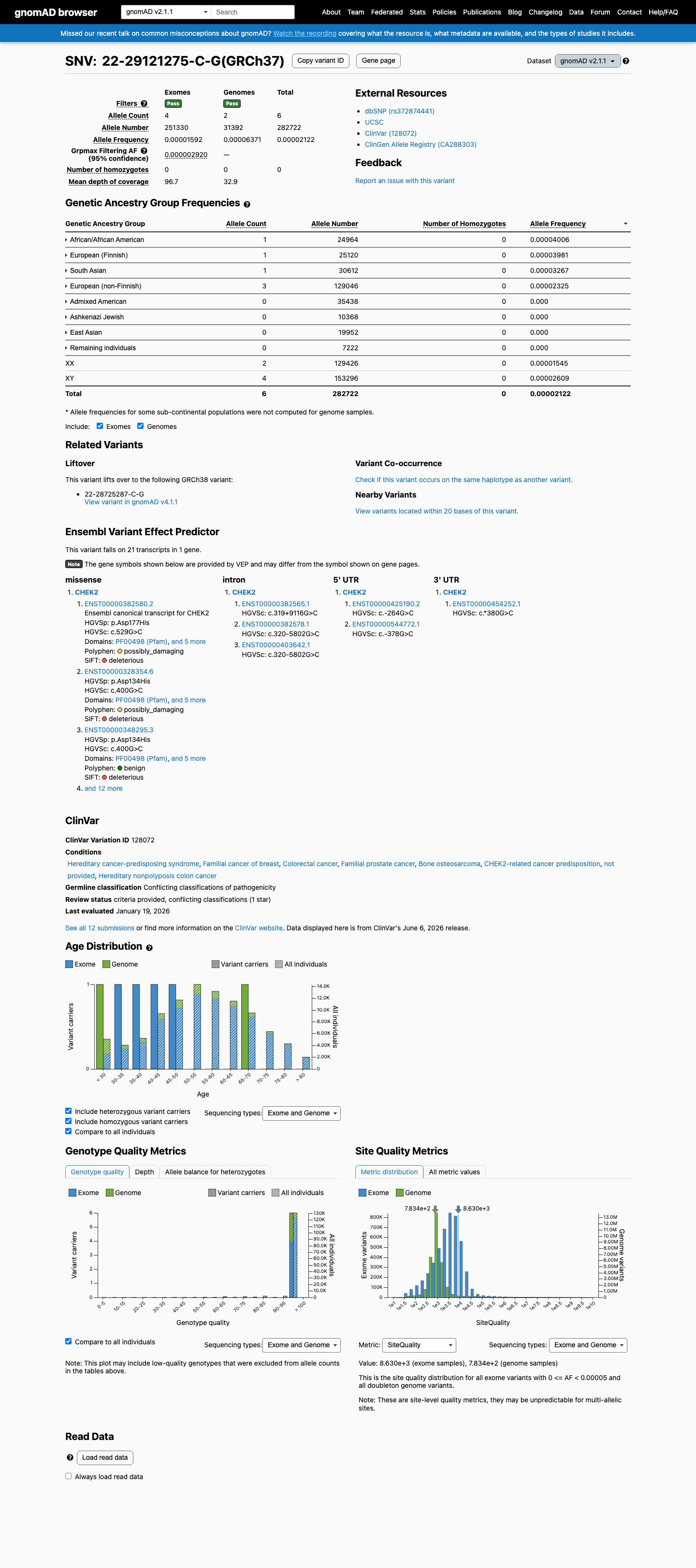
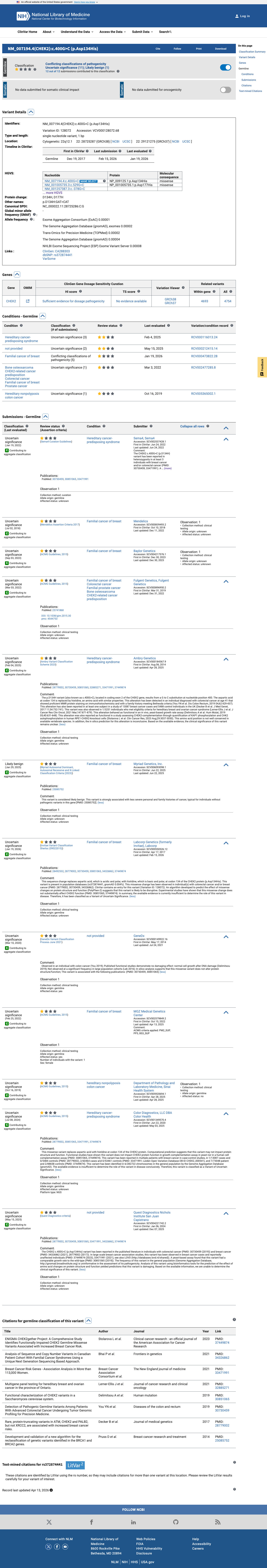
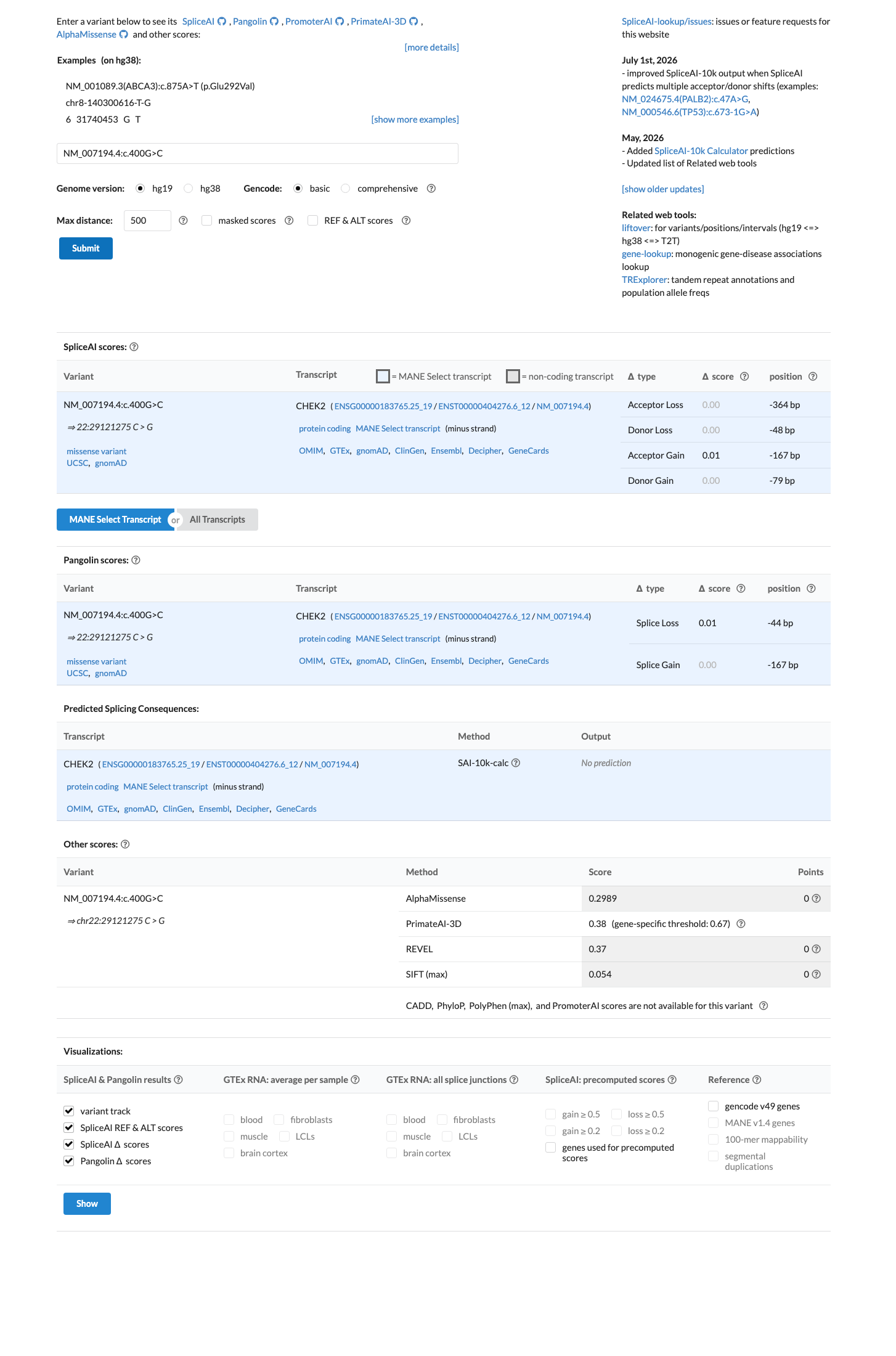
NM_007194.4:c.400G>C (p.Asp134His) in CHEK2 is a rare missense variant observed at extremely low frequency in population databases (gnomAD v2.1 AF=0.00212%, 6/282,722 alleles), meeting PM2 (supporting).1 Multiple in silico predictors are consistent with a benign effect: REVEL score 0.37, BayesDel score -0.100, and SpliceAI max delta 0.01 (no predicted splicing impact), meeting BP4 (supporting).2 No variant-specific functional evidence, case-control data, segregation data, or de novo reports were identified in the literature. The ENIGMA CHEK2gether functional study (PMID:37449874) assessed 430 CHEK2 missense VUS but did not include this variant.3 ClinVar reports this variant as Uncertain significance (10 clinical laboratories) with one Likely benign submission. No expert panel classification is available.4 With one supporting pathogenic criterion (PM2) and one supporting benign criterion (BP4), the evidence is insufficient to classify this variant as either pathogenic or benign. The variant is classified as a Variant of Uncertain Significance (VUS).5
CHEK2
Final classification
VUS
CHEK2 c.400G>C · p.Asp134His
CHEK2
NM_007194.4:c.400G>C (p.Asp134His) in CHEK2 is a rare missense variant observed at extremely low frequency in population databases (gnomAD v2.1 AF=0.00212%, 6/282,722 alleles), meeting PM2 (supporting).
Hereditary Breast, Ovarian and Pancreatic Cancer Specification v1.0.0 lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting benign; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP4
VUS
CHEK2 c.400G>C
PM2 + BP4
→
VUS
2
revelbayesdelspliceai ↗
5
PMID:25741868 ↗generic_acmg_combination_rules
Gene diagram
· NM_007194.4 · variants mapped to exon structure
CHEK2
NM_007194.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 18 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is extremely rare in population databases. In gnomAD v2.1, it is observed at AF=2.12×10⁻⁵ (6/282,722 alleles, 0 homozygotes) and in gnomAD v4.1 at AF=1.12×10⁻⁵ (18/1,613,968 alleles, 0 homozygotes). The highest subpopulation frequency is 4.01×10⁻⁵ in African/African American. These frequencies are well below the PM2 threshold of 0.1%.
gnomAD v2.1 AF=0.00212% (6/282722)v4.1 AF=0.00112% (18/1613968)no homozygotes
✓
BP4
supporting
Benign
Multiple independent in silico predictors are consistent with a benign or neutral effect. REVEL score is 0.37 (below the typical pathogenic threshold of 0.5). BayesDel score is -0.100 (negative, consistent with benign). SpliceAI max delta score is 0.01 (predicts no splicing impact). The concordance of three orthogonal computational tools supports a lack of functional effect.
REVEL=0.37BayesDel=-0.100SpliceAI=0.01 — all consistent with no deleterious effect
Assessed · not applied
Pathogenic
PS1
No known pathogenic variant at the same amino acid position (p.Asp134) with a different nucleotide change was identified in ClinVar or the literature.
PS2
No de novo occurrence data for NM_007194.4:c.400G>C was identified in ClinVar, the reviewed literature, or any other source.
PS3
No variant-specific functional evidence supporting a damaging effect was identified.
PS4
No case-control enrichment data or case series specific to this variant was identified.
PM1
The p.Asp134 residue does not lie in a statistically significant mutational hotspot.
PM5
No pathogenic missense variant at the same amino acid residue (p.Asp134) with a different amino acid change was identified.
PP1
No co-segregation data is available for this variant.
PP2
CHEK2 is a cancer predisposition gene in which both truncating and missense variants are established disease mechanisms.
PP3
Multiple in silico predictors do not support a deleterious effect.
PP4
No specific phenotype data or clinical presentation details for individuals carrying this variant were identified in the reviewed literature or ClinVar submissions.
Benign
BA1
The allele frequency is far below the BA1 threshold of 1%.
BS1
The allele frequency is below the BS1 threshold of 0.3%.
BS2
No homozygous observations in gnomAD (0 homozygotes in both v2.1 and v4.1).
BS3
No variant-specific functional evidence demonstrating a benign effect was identified.
BS4
No segregation data is available for this variant.
BP2
No observation of this variant in trans with a known pathogenic CHEK2 variant was identified.
BP5
No observation of this variant in a case with an alternative molecular basis for disease was identified in the reviewed data.
BP6
No reputable source has classified this variant as benign.
N/A · 8
PVS1 · PM3 · PM4 · PM6 · PP5 · BP1 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 1.11526e-05; MAF= 0.00112%, 18/1613968 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 2.19582e-05; MAF= 0.00220%, 2/91082 alleles, homozygotes = 0); grpmax FAF= 6.15e-06.
v2.1
This variant is present in gnomAD v2.1 (AF= 2.12223e-05; MAF= 0.00212%, 6/282722 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 4.00577e-05; MAF= 0.00401%, 1/24964 alleles, homozygotes = 0); grpmax FAF= 2.92e-06.
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0011%
· 18 / 1,613,968
0 hom · FAF 0.00062%
0 hom · FAF 0.00062%
South Asian 2 / 91,082 |
0.0022% |
Admixed American 1 / 59,974 |
0.0017% |
European (Finnish) 1 / 64,038 |
0.0016% |
African/African American 1 / 74,932 |
0.0013% |
European (non-Finnish) 13 / 1,180,000 |
0.0011% |
+ 5 not observed (Remaining individuals, Amish, East Asian, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.0021%
· 6 / 282,722
0 hom · FAF 0.00029%
0 hom · FAF 0.00029%
African/African American 1 / 24,964 |
0.004% |
European (Finnish) 1 / 25,120 |
0.004% |
South Asian 1 / 30,612 |
0.0033% |
European (non-Finnish) 3 / 129,046 |
0.0023% |
+ 4 not observed (Admixed American, Ashkenazi Jewish, East Asian, Remaining individuals)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Uncertain significance (10 clinical laboratories) and as Likely benign (1 clinical laboratory). (ClinVarID = 128072)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). REVEL score = 0.37. BayesDel score = -0.100278.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. CHEK2, an intracellular kinase involved in control of the cell cycle, is altered in various cancer types.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
9Sources
Triaged references · 9 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
28779002 ↗
Rare, protein-truncating variants in ATM, CHEK2 and PALB2, but not XRCC2, are associated with increased breast cancer risks.
CLINVAR
30730459 ↗
Detection of Pathogenic Germline Variants Among Patients With Advanced Colorectal Cancer Undergoing Tumor Genomic Profiling for Precision Medicine.
CLINVAR
30851065 ↗
Functional characterization of CHEK2 variants in a Saccharomyces cerevisiae system.
CLINVAR
34326862 ↗
Analysis of Sequence and Copy Number Variants in Canadian Patient Cohort With Familial Cancer Syndromes Using a Unique Next Generation Sequencing Based Approach.
CLINVAR
37449874 ↗
ENIGMA CHEK2gether Project: A Comprehensive Study Identifies Functionally Impaired CHEK2 Germline Missense Variants Associated with Increased Breast Cancer Risk.
CLINVAR
15604628 ↗
Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR