PM1 (moderate) is met: the variant alters Ile251 in the CHEK2 kinase domain, a well-established critical functional domain.1 PM2 (supporting) is met: the variant is present at extremely low frequency in population databases (gnomAD v2.1 AF=0.00177%, gnomAD v4.1 AF=0.00328%).2 BS4 (supporting benign) is met: the variant demonstrated lack of segregation in a familial prostate cancer family, where it was detected in only 1 of 2 affected brothers.3 The variant has been observed in individual cases of familial prostate cancer (1/298) and hereditary breast cancer (1/507) but has not demonstrated statistically significant case-control enrichment.4 No variant-specific functional studies have been performed for p.Ile251Phe. In silico predictions are mixed: REVEL=0.426, BayesDel=0.178, PolyPhen-2=Probably damaging, SIFT=Tolerated.5 ClinVar classification is Uncertain significance (15 VUS, 3 Likely pathogenic submissions). No expert panel review has been performed.6 With one moderate pathogenic criterion (PM1), one supporting pathogenic criterion (PM2), and one supporting benign criterion (BS4), the evidence is equivocal and the variant remains a Variant of Uncertain Significance (VUS).7
CHEK2
Final classification
VUS
CHEK2 c.751A>T · p.Ile251Phe
CHEK2
PM1 (moderate) is met: the variant alters Ile251 in the CHEK2 kinase domain, a well-established critical functional domain.
Hereditary Breast, Ovarian and Pancreatic Cancer Specification v1.0.0 lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM1 moderate, PM2 supporting, BS4 supporting benign; combination = 1 moderate + 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM1PM2
BS4
VUS
CHEK2 c.751A>T
PM1 + PM2 + BS4
→
VUS
5
PMID:22114986 ↗revelbayesdelspliceai ↗
7
generic_acmg_combination_rules
Gene diagram
· NM_007194.4 · variants mapped to exon structure
CHEK2
NM_007194.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 21 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PM1
moderate
Pathogenic
The variant alters residue Ile251 in the CHEK2 kinase domain (residues ~220–500), a well-established critical functional domain essential for CHEK2 kinase activity and DNA damage signaling. Missense substitutions in the kinase domain have been shown to abolish CHEK2 function.
Located in the kinase domain of CHEK2 (exon 6). The kinase domain is a well-established critical functional domain involved in phosphorylation of downstream targets including p53 and BRCA1.
✓
PM2
supporting
Pathogenic
The variant is present at an extremely low frequency in population databases: gnomAD v2.1 AF=0.00177% (5/282,756 alleles), gnomAD v4.1 AF=0.00328% (53/1,613,926 alleles), and absent from gnomAD-Canada. These frequencies are well below the PM2 threshold of 0.1%.
gnomAD v2.1: 5/282756 alleles (AF=0.00177%)gnomAD v4.1: 53/1
✓
BS4
supporting
Benign
In the familial prostate cancer family reported by Dong et al. (PMID:12533788), the c.751A>T variant was detected in only 1 of 2 affected brothers, demonstrating lack of segregation with disease. This represents a non-segregation event in affected family members supporting a benign interpretation.
Variant found in only 1 of 2 affected brothers in a familial prostate cancer family (PMID:12533788)indicating lack of segregation.
Assessed · not applied
Pathogenic
PVS1
This variant is a missense substitution (c.751A>T, p.Ile251Phe) and does not fall into a null-variant category (nonsense, frameshift, canonical ±1,2 splice site, initiation codon, or exon deletion).
PS1
No evidence that a different nucleotide change at c.751 resulting in the same amino acid alteration (p.Ile251Phe) has been established as pathogenic.
PS2
No de novo observation has been reported for this variant.
PS3
No variant-specific functional assay has been performed for p.Ile251Phe.
PS4
The variant has been observed in individual cases across two studies (1/298 familial prostate cancer in Dong et al.
PM5
No pathogenic missense variant at the same amino acid residue (Ile251) has been identified in ClinVar or the literature.
PM6
No de novo observation has been reported for this variant.
PP1
In the familial prostate cancer family reported by Dong et al.
PP2
Gene-level missense constraint data are not available to confirm that CHEK2 has a low rate of benign missense variation.
PP3
Computational evidence is mixed and does not meet the threshold for multiple lines supporting a deleterious effect.
PP4
The variant has been observed in patients with prostate cancer, breast cancer, and colorectal cancer — phenotypes that are not highly specific for a single genetic etiology.
PP5
The variant is classified as 'Uncertain significance' overall in ClinVar (ClinVarID 128086).
Benign
BA1
The maximum population allele frequency is 0.00387% (gnomAD v2.1, European non-Finnish), far below the BA1 threshold of 1%.
BS1
The population allele frequency (gnomAD v2.1 AF=0.00177%, gnomAD v4.1 AF=0.00328%) is well below the BS1 threshold of 0.3%.
BS2
No homozygous individuals have been observed, and there are no confirmed observations of this variant in healthy adult individuals with documented normal phenotype to satisfy BS2.
BS3
No variant-specific functional assay demonstrating a benign effect has been performed for p.Ile251Phe.
BP1
CHEK2 has multiple established pathogenic missense variants (e.g., p.Ile157Thr, p.Arg145Trp, p.Arg117Gly, p.Thr476Met) in addition to truncating variants.
BP2
No evidence of this variant being observed in trans with a known pathogenic CHEK2 variant, or in cis with a known pathogenic variant in any inheritance pattern.
BP4
Computational evidence does not converge on a benign prediction.
BP5
No evidence that this variant has been found in a case with an alternate molecular basis for disease.
BP6
No reputable source has classified this variant as benign.
N/A · 4
PM3 · PM4 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
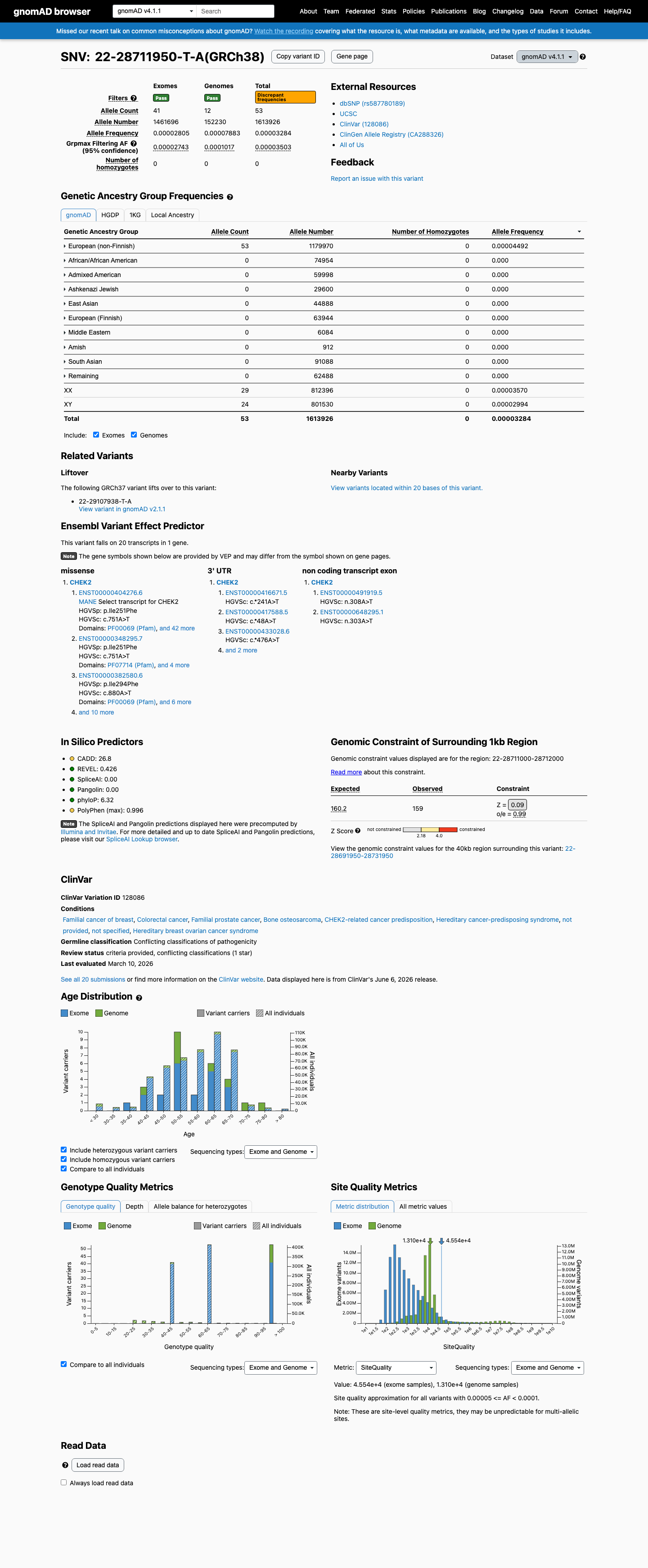
This variant is present in gnomAD v4.1 (AF= 3.28392e-05; MAF= 0.00328%, 53/1613926 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 4.49164e-05; MAF= 0.00449%, 53/1179970 alleles, homozygotes = 0); grpmax FAF= 3.503e-05.
v2.1
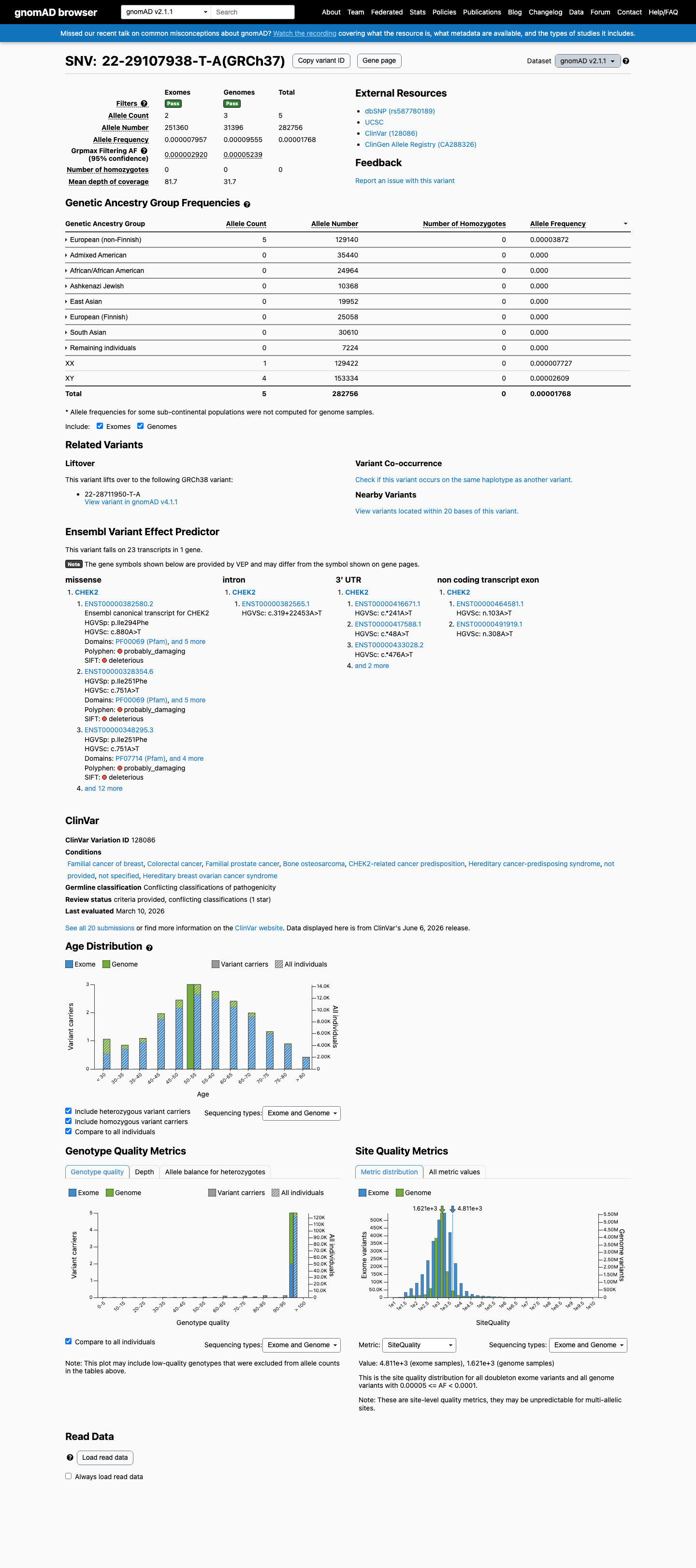
This variant is present in gnomAD v2.1 (AF= 1.76831e-05; MAF= 0.00177%, 5/282756 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 3.87177e-05; MAF= 0.00387%, 5/129140 alleles, homozygotes = 0); grpmax FAF= 5.239e-05.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.00016286644951140066, 3/18420 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0033%
· 53 / 1,613,926
0 hom · FAF 0.0035%
0 hom · FAF 0.0035%
European (non-Finnish) 53 / 1,179,970 |
0.0045% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0018%
· 5 / 282,756
0 hom · FAF 0.0052%
0 hom · FAF 0.0052%
European (non-Finnish) 5 / 129,140 |
0.0039% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
0.016%
· 3 / 18,420
0 hom · FAF 0.0069%
0 hom · FAF 0.0069%
European (non-Finnish) 3 / 11,742 |
0.026% |
+ 8 not observed (African/African American, Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, Remaining individuals, South Asian)
ClinVar
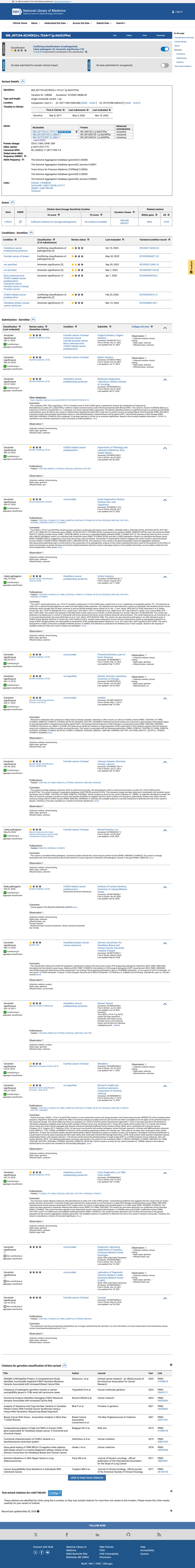
This variant has been reported in ClinVar as Uncertain significance (15 clinical laboratories) and as Likely pathogenic (2 clinical laboratories) and as likely pathogenic (1 clinical laboratory). (ClinVarID = 128086)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.426. BayesDel score = 0.177609.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. CHEK2, an intracellular kinase involved in control of the cell cycle, is altered in various cancer types.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 6 further PMIDs triaged but not cited — see Sources & References.
Mutations in CHEK2 associated with prostate cancer risk.
Searched
c.751A>TA751TIle251PheI251F751A>T
Found
A751T (Ile251Phe) identified as a germline CHEK2 missense mutation in 1 of 298 familial prostate cancer probands and absent in 423 unaffected controls. The variant was found in only 1 of 2 affected brothers in the family tested for segregation. The missense variant was not individually characterized in functional assays; western blot analysis of pooled missense mutations showed normal CHEK2 protein levels.
Variant
✓ Names this variant — characterised directly
Applied to
→BS4 supports · met
→PM1 supports · met
Why
Variant specifically identified in a familial prostate cancer case. Provides evidence for PM1 (kinase domain location) and BS4 (non-segregation in affected siblings).
three were missense mutations—one in exon 3 (T470C, Ile157Thr) and two in exon 5 (G715A, Glu239Lys and A751T, Ile251Phe)
Location Table 1 (mutation #13); Results paragraphs 1, 3, 5; Figure 3C · Context DHPLC mutation screening followed by Sanger sequencing; western blot analysis in EBV-transformed lymphoblastoid cell lines · full text
CHEK2 contribution to hereditary breast cancer in non-BRCA families.
Searched
c.751A>T751A>TIle251PheI251F
Found
751A>T (Ile251Phe) identified in 1 of 507 non-BRCA hereditary breast cancer cases and absent in 513 controls. In silico analysis: Align-GVGD class C0, SIFT tolerated (score 2.04), PolyPhen-2 probably damaging. Classified as 'probably deleterious' by study criteria. This variant was not included in the in vitro kinase activity assay.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
Why
Variant identified in a hereditary breast cancer case with mixed in silico predictions. Not functionally characterized. Supports PM1 (kinase domain).
751A > T [ 23 ] Ile251Phe 6 Kinase - C0 2.04 Tolerated Probably damaging Probably deleterious
Location Table 1 (case population mutations); Results paragraph 1 · Context In silico prediction only; variant was not among the 11 variants tested in the Omnia in vitro kinase activity assay using recombinant Flag-CHEK2 expressed in E. coli · full text
Sources & reference links
9Sources
Triaged references · 6 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
27498913 ↗
Monogenic and polygenic determinants of sarcoma risk: an international genetic study.
CLINVAR
27978560 ↗
Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer.
CLINVAR