Classification rationale
BS1BS3BP1
Benign
BRCA1 c.425C>A
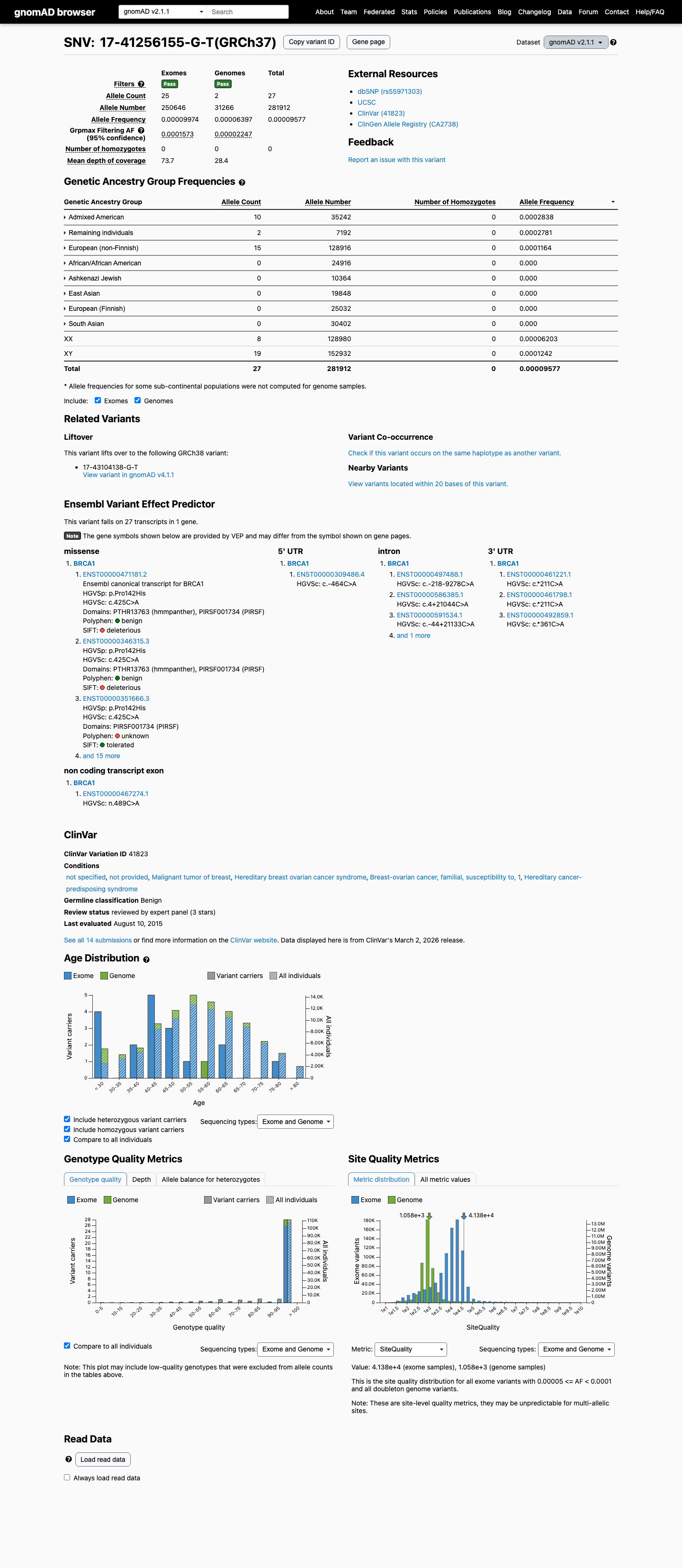
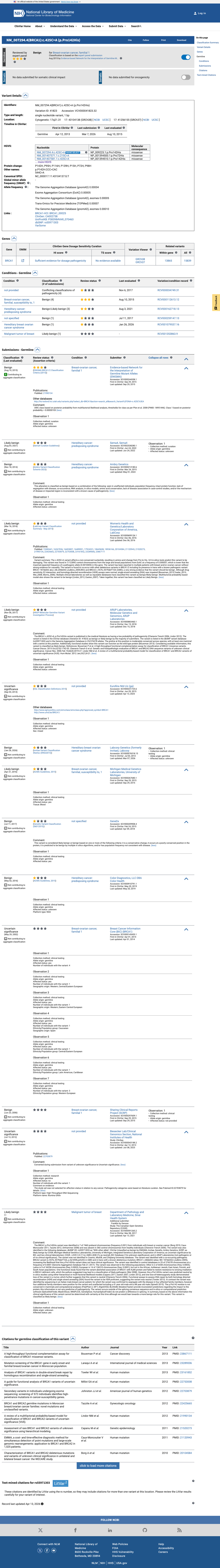
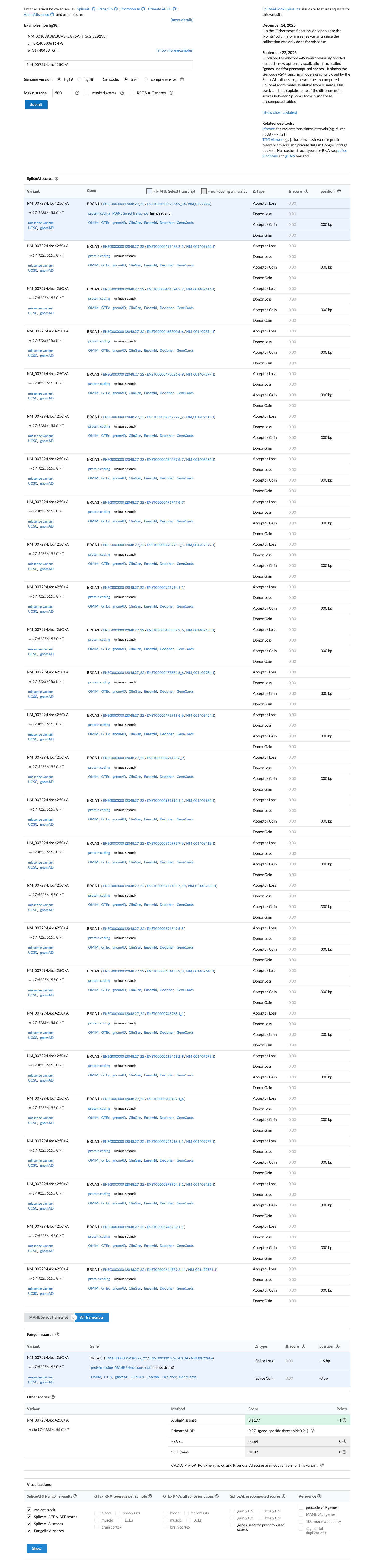
The BRCA1 c.425C>A (p.Pro142His) variant has been reported in ClinVar with an ENIGMA expert panel benign classification.1 This variant is present in gnomAD, and the highest observed filter allele frequencies exceed the ENIGMA BS1_Strong threshold of 0.0001 (gnomAD v2.1 grpmax FAF 0.00015726; gnomAD v4.1 joint grpmax FAF 0.00015364).2 Calibrated BRCA1 functional evidence supports no damaging effect, with ENIGMA Table 9 assigning BS3_Strong and supplementary functional data describing no functional impact.3 This missense change is outside the BRCA1 clinically important functional domains, and SpliceAI predicts no significant splice effect with a maximum delta score of 0.00, which is consistent with BP1_Strong and does not support PP3.4
BS1 + BS3 + BP1
→
Benign
3
vcep_s_p_e_c_i_f_i_c_a_t_i_o_n_s___t_a_b_l_e_9___v_1___2___2_0_2_4___1_1___1_8vcep_s_u_p_p_l_e_m_e_n_t_a_r_y_t_a_b_l_e_s___v_1___2___2_0_2_4___1_1___1_8
4
cspec ↗spliceai ↗vcep_a_p_p_e_n_d_i_c_e_s___v_1___2___2_0_2_4___1_1___1_8