NM_014727.2:c.5989C>G (p.Leu1997Val) in KMT2B is extremely rare in population databases, observed in only 1 of 1,613,902 alleles (AF=0.00006%) in gnomAD v4.1 and absent from gnomAD v2.1 and gnomAD-Canada, meeting PM2 at supporting strength.1 This variant is a missense substitution and does not qualify for PVS1; no pathogenic variants have been reported at the same residue (PS1 not met); no functional studies exist (PS3 not assessed); and no de novo or segregation data are available. Multiple in silico prediction tools do not support a deleterious effect: REVEL score is 0.37 (intermediate), BayesDel score is -0.166 (benign range), and SpliceAI predicts no splicing impact (max delta=0.00). However, this evidence is mixed and does not confidently meet PP3 or BP4.2 The only criterion met is PM2 (supporting). Per ACMG/AMP 2015 classification rules (PMID:25741868), a single supporting pathogenic criterion without any benign criteria is insufficient for a likely pathogenic or benign classification.3 Overall classification: Variant of Uncertain Significance (VUS). Additional evidence including functional studies, segregation analysis, case-control data, or clinical correlation is required to resolve the significance of this variant.
KMT2B
Final classification
VUS
KMT2B c.5989C>G · p.Leu1997Val
KMT2B
NM_014727.2:c.5989C>G (p.Leu1997Val) in KMT2B is extremely rare in population databases, observed in only 1 of 1,613,902 alleles (AF=0.00006%) in gnomAD v4.1 and absent from gnomAD v2.1 and gnomAD-Canada, meeting PM2 at supporting strength.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting; combination = 1 supporting, which maps to VUS.
Classification rationale
PM2
VUS
KMT2B c.5989C>G
PM2
→
VUS
Gene diagram
· NM_014727.2 · variants mapped to exon structure
KMT2B
NM_014727.2
Fetching transcript structure from UCSC…
Applied criteria · 1 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
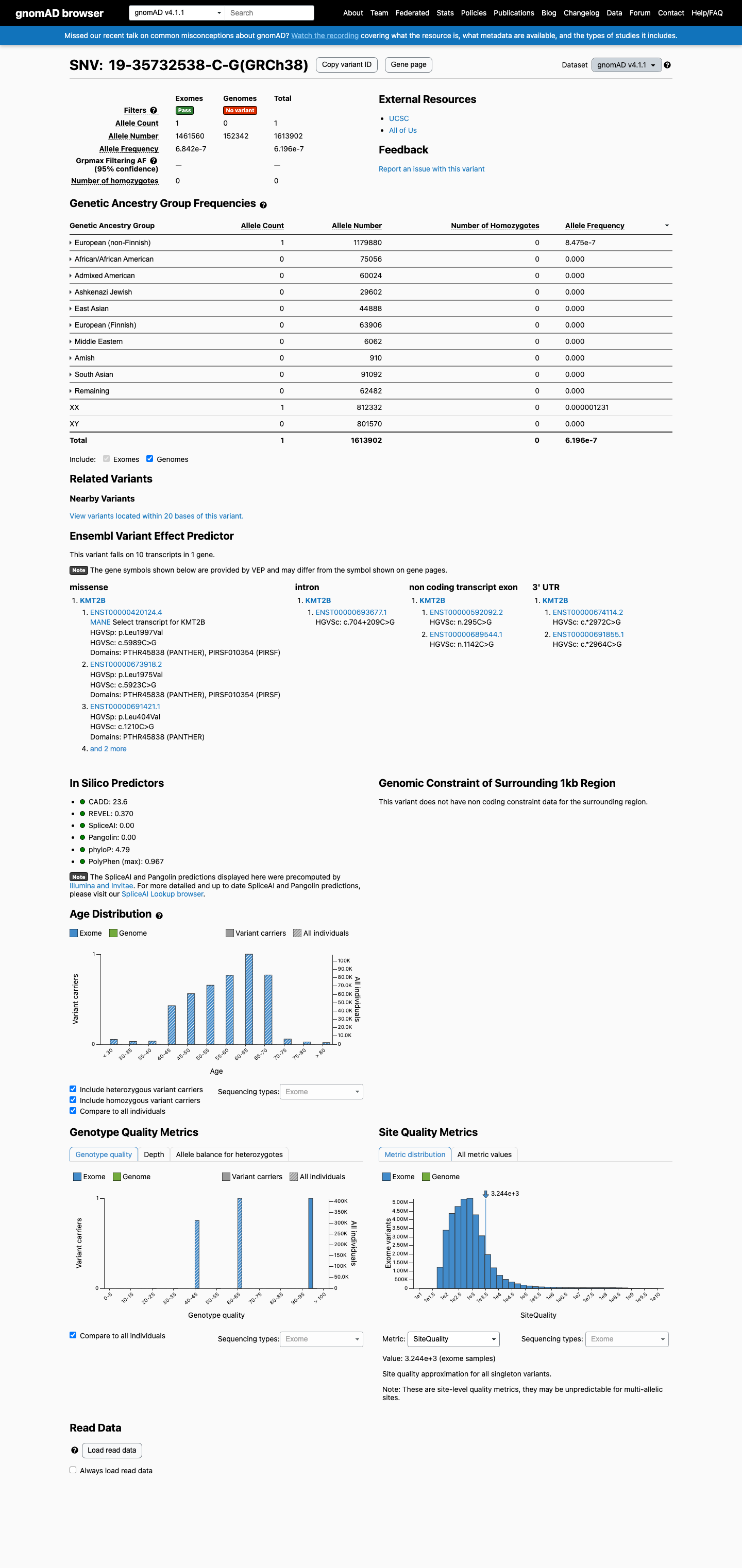
This variant is present in gnomAD v4.1 (AF= 6.19616e-07; MAF= 0.00006%, 1/1613902 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.47544e-07; MAF= 0.00008%, 1/1179880 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
6.2e-05%
· 1 / 1,613,902
0 hom
0 hom
European (non-Finnish) 1 / 1,179,880 |
8.5e-05% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.37. BayesDel score = -0.166046.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. KMT2B, a histone methyltransferase, is mutated at low frequencies in various cancer types.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links