NM_016507.4:c.2964C>T (p.Phe988=) is a synonymous variant in CDK12. This variant is present in gnomAD at a grpmax filtering allele frequency of 0.798% (v2.1) and 0.781% (v4.1), with an African/African American subpopulation frequency of 0.885%, meeting BS1 at strong benign strength.1 The variant is observed in 2 homozygous individuals in gnomAD v2.1 and 4 homozygous individuals in gnomAD v4.1, further supporting that biallelic presence is tolerated in the general population.2 SpliceAI predicts no impact on splicing (max delta score 0.02), consistent with a synonymous variant that does not alter the protein product or disrupt normal mRNA processing, meeting BP7 at supporting benign strength.3 Computational splicing predictions show no evidence of splice site disruption, meeting BP4 at supporting benign strength.4 ClinVar reports this variant as Likely benign (2 clinical laboratories) and Benign (1 clinical laboratory); however the review status is 1-star (criteria provided, single submitter) and does not independently meet the 3-star expert panel threshold for BP6.5 No pathogenic criteria are met. BS1 (strong benign), BP4 (supporting benign), and BP7 (supporting benign) together support a classification of Likely benign per ACMG/AMP 2015 guidelines.6
CDK12
Final classification
Likely Benign
CDK12 c.2964C>T · p.Phe988=
CDK12
NM_016507.4:c.2964C>T (p.Phe988=) is a synonymous variant in CDK12. This variant is present in gnomAD at a grpmax filtering allele frequency of 0.798% (v2.1) and 0.781% (v4.1), with an African/African American subpopulation frequency of 0.885%, meeting BS1 at strong benign strength.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS1 strong benign, BP4 supporting benign, BP7 supporting benign; combination = 1 strong benign + 2 supporting benign, which maps to Likely Benign.
Classification rationale
BS1BP4BP7
Likely Benign
CDK12 c.2964C>T
BS1 + BP4 + BP7
→
Likely Benign
Gene diagram
· NM_016507.4 · variants mapped to exon structure
CDK12
NM_016507.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 18 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
strong
Benign
The allele frequency of NM_016507.4:c.2964C>T in population databases exceeds the threshold expected for a pathogenic CDK12 variant. In gnomAD v2.1, the variant has a grpmax filtering allele frequency of 0.798% (African/African American subpopulation AF 0.885%) with 2 homozygous individuals. In gnomAD v4.1, the grpmax FAF is 0.781% (African/African American AF 0.835%) with 4 homozygous individuals. These frequencies are well above the BS1 threshold of >0.3% for a rare Mendelian disorder gene, supporting that this variant is too common in the general population to be pathogenic.
gnomAD v2.1: total AF 0.086% (243/282676 alleles)African/African American AF 0.885% (221/24
✓
BP4
supporting
Benign
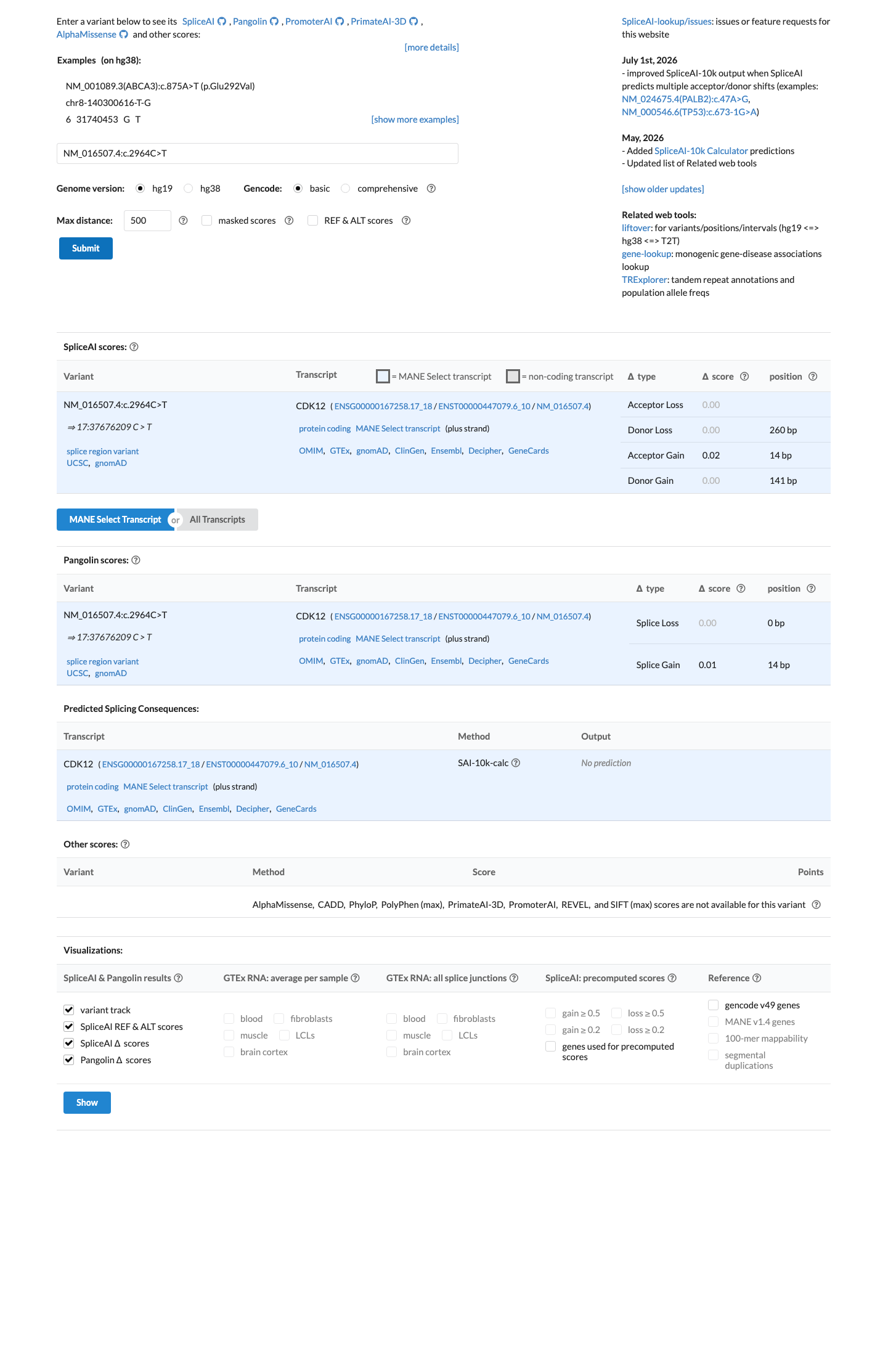
Computational evidence predicts no impact on splicing for this synonymous variant. SpliceAI assigns a maximum delta score of 0.02 (well below the 0.2 threshold), with no predicted donor gain, donor loss, acceptor gain, or acceptor loss. This supports a benign interpretation.
SpliceAI max delta score 0.02: no donor/acceptor gain or lossno significant splicing impact predicted.
✓
BP7
supporting
Benign
NM_016507.4:c.2964C>T is a synonymous variant (p.Phe988=) with SpliceAI predicting no splice impact (max delta score 0.02). No donor/acceptor gain or loss is predicted. The nucleotide substitution is not at a highly conserved splice consensus position and computational predictions support no effect on splicing. BP7 is met at supporting benign strength.
Synonymous variant producing p.(Phe988=).SpliceAI max delta score 0.02: no donor or acceptor site gain or loss predicted.Nucleotide substitution is not within the splice consensus region.
Assessed · not applied
Pathogenic
PS1
PS1 requires a different nucleotide change at the same codon producing the same missense change that has been previously classified as pathogenic.
PS2
No evidence of de novo occurrence for NM_016507.4:c.2964C>T in any published study or database.
PS3
No functional data identified for this synonymous variant.
PS4
No case-control studies demonstrating enrichment of this variant in affected individuals compared to controls.
PM1
Although CDK12 contains a kinase domain (approximately residues 723-1002) and position 988 falls within the C-terminal region of this domain, this is a synonymous variant (p.Phe988=) with no predicted splice impact (SpliceAI max delta 0.02).
PM2
This variant is present in gnomAD population databases at a frequency exceeding the PM2 threshold of <0.1%.
PM6
No evidence of de novo occurrence for NM_016507.4:c.2964C>T.
PP1
No cosegregation data available for NM_016507.4:c.2964C>T with disease in affected family members.
PP3
In silico prediction tools do not support a deleterious effect.
PP4
No evidence that the patient's phenotype or family history is highly specific for CDK12-related disease.
PP5
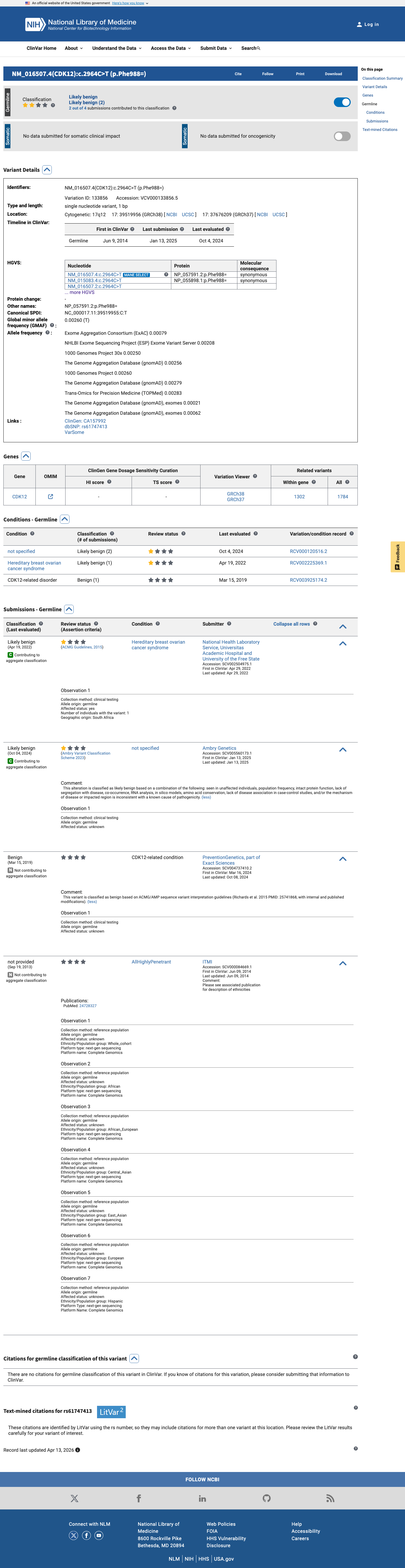
ClinVar lists this variant as 'Likely benign' (ClinVar Variation ID 133856) with review status 'criteria provided, single submitter' (1-star).
Benign
BA1
The maximum subpopulation allele frequency in gnomAD is 0.885% in the African/African American population (gnomAD v2.1), with a grpmax FAF of 0.798%.
BS2
Although the variant is observed in 2 homozygous individuals in gnomAD v2.1 and 4 homozygous individuals in gnomAD v4.1, BS2 requires observation in a healthy adult for a disorder with full penetrance expected at an early age.
BS3
No functional studies demonstrating a neutral (benign) effect for NM_016507.4:c.2964C>T.
BS4
No cosegregation data available demonstrating lack of segregation with disease for NM_016507.4:c.2964C>T.
BP2
No evidence of this variant observed in trans with a known pathogenic variant in CDK12.
BP5
No evidence that this variant is found in a case with an alternate molecular basis for disease.
BP6
ClinVar reports this variant as 'Likely benign' (Variation ID 133856) with review status 'criteria provided, single submitter' (1-star).
N/A · 4
PVS1 · PM5 · PP2 · BP1
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
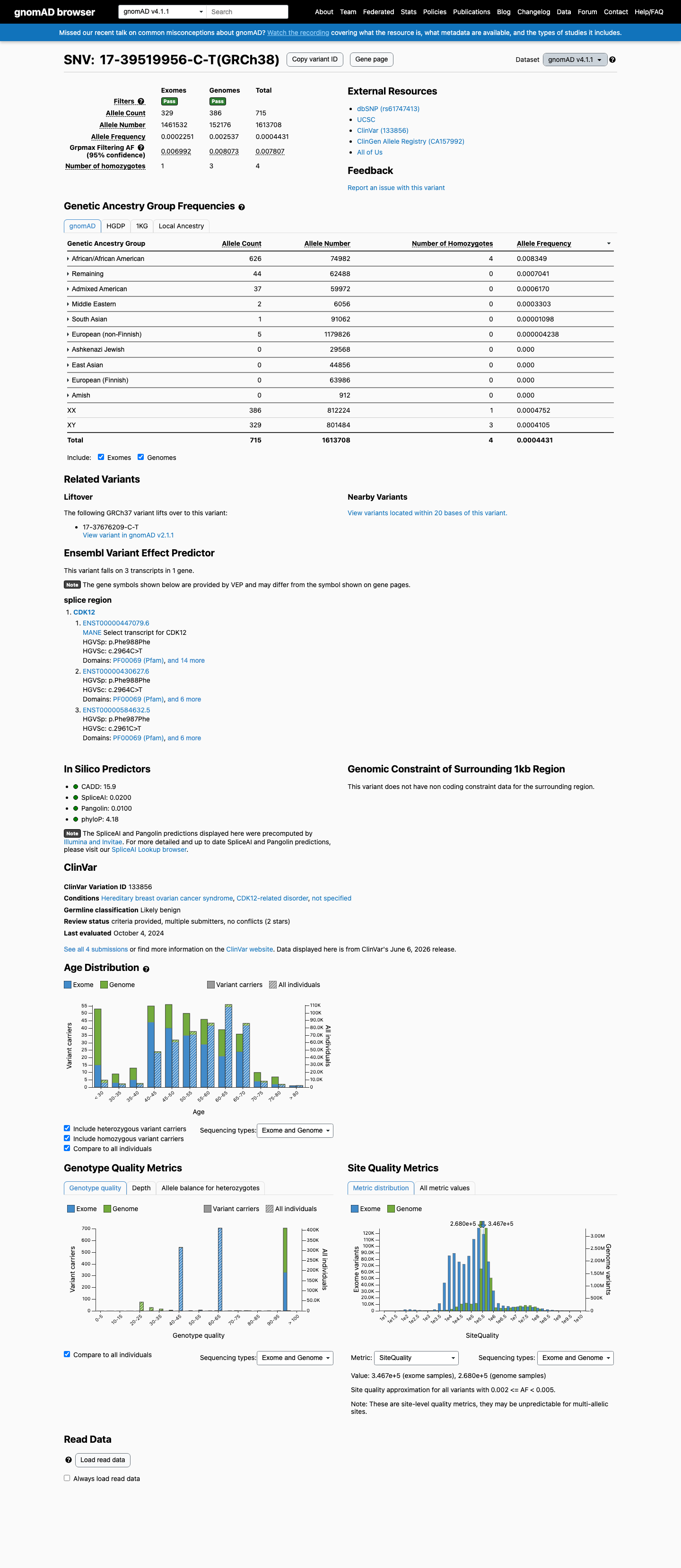
This variant is present in gnomAD v4.1 (AF= 0.000443079; MAF= 0.04431%, 715/1613708 alleles, homozygotes = 4) and has highest observed frequency in the African/African American population (AF= 0.00834867; MAF= 0.83487%, 626/74982 alleles, homozygotes = 4); grpmax FAF= 0.00780671.
v2.1
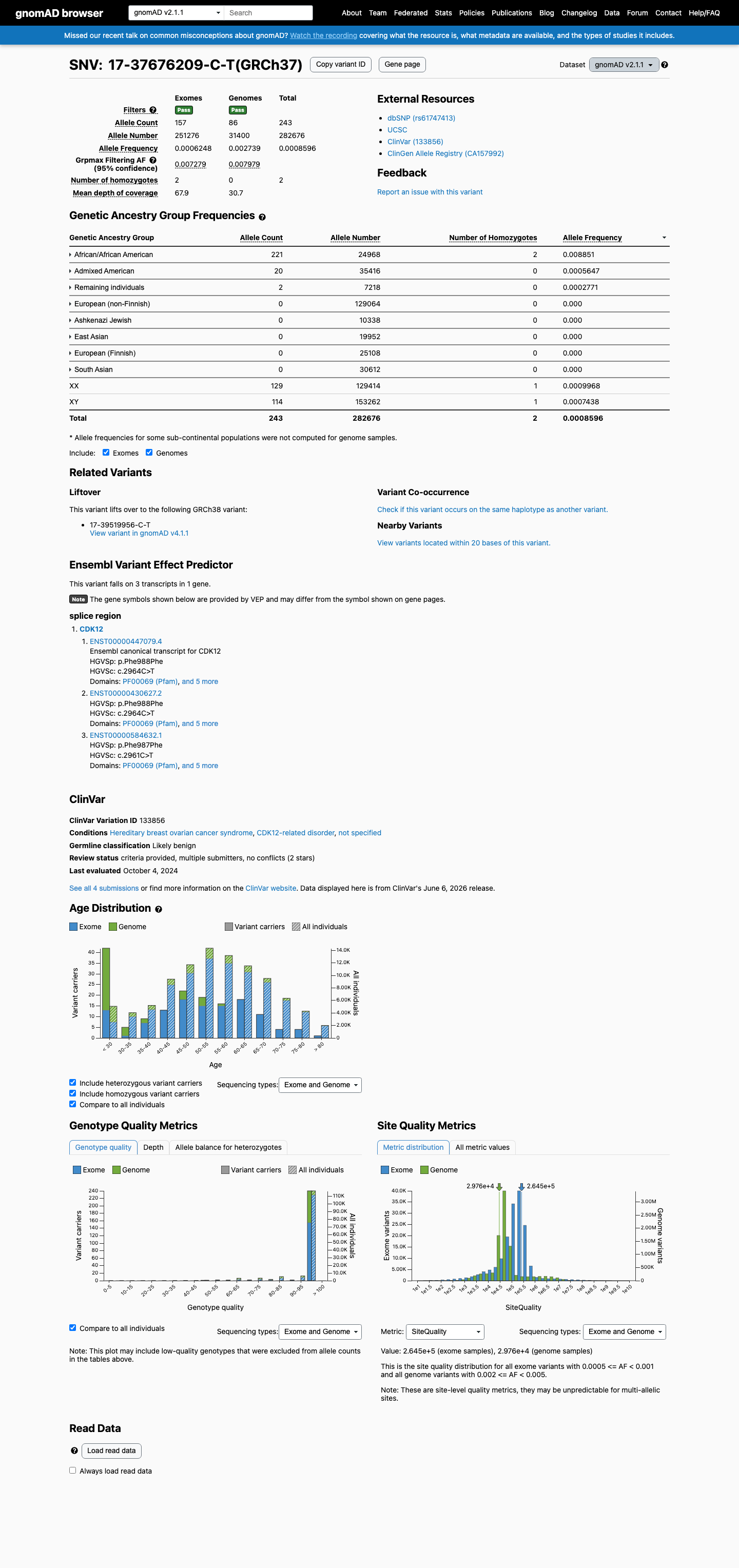
This variant is present in gnomAD v2.1 (AF= 0.000859641; MAF= 0.08596%, 243/282676 alleles, homozygotes = 2) and has highest observed frequency in the African/African American population (AF= 0.00885133; MAF= 0.88513%, 221/24968 alleles, homozygotes = 2); grpmax FAF= 0.00797854.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0008686210640608035, 16/18420 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.044%
· 715 / 1,613,708
4 hom · FAF 0.78%
4 hom · FAF 0.78%
African/African American 626 / 74,982 |
0.83% 4 hom |
Remaining individuals 44 / 62,488 |
0.07% |
Admixed American 37 / 59,972 |
0.062% |
Middle Eastern 2 / 6,056 |
0.033% |
South Asian 1 / 91,062 |
0.0011% |
European (non-Finnish) 5 / 1,179,826 |
0.00042% |
+ 4 not observed (European (Finnish), Amish, East Asian, Ashkenazi Jewish)
gnomAD v2.1
0.086%
· 243 / 282,676
2 hom · FAF 0.8%
2 hom · FAF 0.8%
African/African American 221 / 24,968 |
0.89% 2 hom |
Admixed American 20 / 35,416 |
0.056% |
Remaining individuals 2 / 7,218 |
0.028% |
+ 5 not observed (Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), South Asian)
gnomAD Canada 🇨🇦
0.087%
· 16 / 18,420
0 hom · FAF 0.98%
0 hom · FAF 0.98%
African/African American 16 / 1,020 |
1.6% |
+ 8 not observed (Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, European (non-Finnish), Remaining individuals, South Asian)
ClinVar
This variant has been reported in ClinVar as Likely benign (2 clinical laboratories) and as Benign (1 clinical laboratory). (ClinVarID = 133856)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02).
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 7 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
17392385 ↗
American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography.
CLINVAR
23188549 ↗
NSGC practice guideline: risk assessment and genetic counseling for hereditary breast and ovarian cancer.
CLINVAR
24366376 ↗
Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women: U.S. Preventive Services Task Force recommendation statement.
CLINVAR
24366402 ↗
Summaries for patients. Assessing the genetic risk for BRCA-related breast or ovarian cancer in women: recommendations from the U.S. Preventive Services Task Force.
CLINVAR
24493721 ↗
American Society of Clinical Oncology Expert Statement: collection and use of a cancer family history for oncology providers.
CLINVAR
24728327 ↗
Germline variation in cancer-susceptibility genes in a healthy, ancestrally diverse cohort: implications for individual genome sequencing.
CLINVAR