NM_016507.4:c.86G>C (p.Ser29Thr) is a missense variant in CDK12 absent from population databases (gnomAD v2.1 and v4.1), supporting PM2 at supporting strength.1 Multiple in silico predictors (REVEL 0.176, BayesDel -0.320, SpliceAI 0.00) support a benign effect, meeting BP4 at supporting strength.2 No functional studies, case-control data, segregation data, de novo reports, ClinVar classifications, or same-residue pathogenic comparators were identified for this variant.3 The variant does not fall within a known functional domain or mutational hotspot, and PVS1 is not applicable as this is a missense substitution.4 Overall, PM2 (supporting) and BP4 (supporting) are the only criteria met, yielding a classification of Variant of Uncertain Significance (VUS) per ACMG/AMP 2015 combination rules — PM2 and BP4 with equal supporting-level evidence in opposing directions do not reach pathogenic or likely benign thresholds.5
CDK12
Final classification
VUS
CDK12 c.86G>C · p.Ser29Thr
CDK12
NM_016507.4:c.86G>C (p.Ser29Thr) is a missense variant in CDK12 absent from population databases (gnomAD v2.1 and v4.1), supporting PM2 at supporting strength.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP4
VUS
CDK12 c.86G>C
PM2 + BP4
→
VUS
2
revelbayesdelspliceai ↗
5
generic_acmg_combination_rules
Gene diagram
· NM_016507.4 · variants mapped to exon structure
CDK12
NM_016507.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 21 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_016507.4:c.86G>C is absent from gnomAD v2.1 and v4.1, consistent with a population frequency below 0.1% (PM2).
Absent from gnomAD v2.1 (exomes)absent from gnomAD v4.1 (exomes).
✓
BP4
supporting
Benign
Multiple lines of in silico evidence predict a benign effect: REVEL score 0.176 (below pathogenic threshold), BayesDel score -0.320 (negative, benign range), and SpliceAI max delta 0.00 (no predicted splicing impact).
REVEL 0.176 (benign range)BayesDel -0.320 (benign range)SpliceAI 0.00 (no splicing impact).
Assessed · not applied
Pathogenic
PS1
No previously established pathogenic variant at the same amino acid position (p.Ser29) has been identified in ClinVar or the literature.
PS2
No de novo data are available for this variant; no reports of confirmed parentage with this variant absent from both parents were identified.
PS3
No functional studies were identified for this variant or for a systematically characterized range that includes p.Ser29.
PS4
No case-control studies or cohort data demonstrating enrichment of this variant in affected individuals versus controls were identified.
PM1
Residue p.Ser29 lies in the N-terminal region of CDK12, far upstream of the kinase domain (residues ~720–980), and is not within a well-characterized critical functional domain.
PM5
No pathogenic variant at the same amino acid residue (p.Ser29) with a different missense change was identified in ClinVar or the literature.
PM6
No de novo event has been reported for this variant with confirmed parentage.
PP1
No segregation data are available for this variant in affected families.
PP2
Insufficient data to confirm that CDK12 has a low rate of benign missense variation and that missense variants are a common mechanism of disease.
PP3
Multiple lines of computational evidence do not support a deleterious effect: REVEL score 0.176 (below 0.5 threshold), BayesDel score -0.320 (negative), and SpliceAI max delta 0.00 (no predicted splicing impact).
PP4
No specific phenotype or clinical data are available for the individual carrying this variant.
PP5
This variant is absent from ClinVar; no pathogenic classification from a reputable source is available.
Benign
BA1
NM_016507.4:c.86G>C is absent from population databases; the allele frequency does not exceed the BA1 threshold of >1%.
BS1
The variant is absent from population databases; the allele frequency does not exceed the BS1 threshold of >0.3%.
BS2
No data are available on healthy adult homozygotes or hemizygotes for this variant.
BS3
No functional studies demonstrating no deleterious effect have been identified for this variant.
BS4
No segregation data are available to demonstrate lack of cosegregation with disease in affected families.
BP1
CDK12 germline disease mechanism is not exclusively mediated by truncating variants; both missense and truncating variants have been reported in association with prostate cancer.
BP2
No data on co-occurrence of this variant in trans with a known pathogenic variant for a fully penetrant dominant disorder.
BP5
No alternate molecular basis for disease has been identified in the case to suggest this variant is not causative.
BP6
This variant is absent from ClinVar; no benign classification from a reputable source is available.
N/A · 5
PVS1 · PM3 · PM4 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

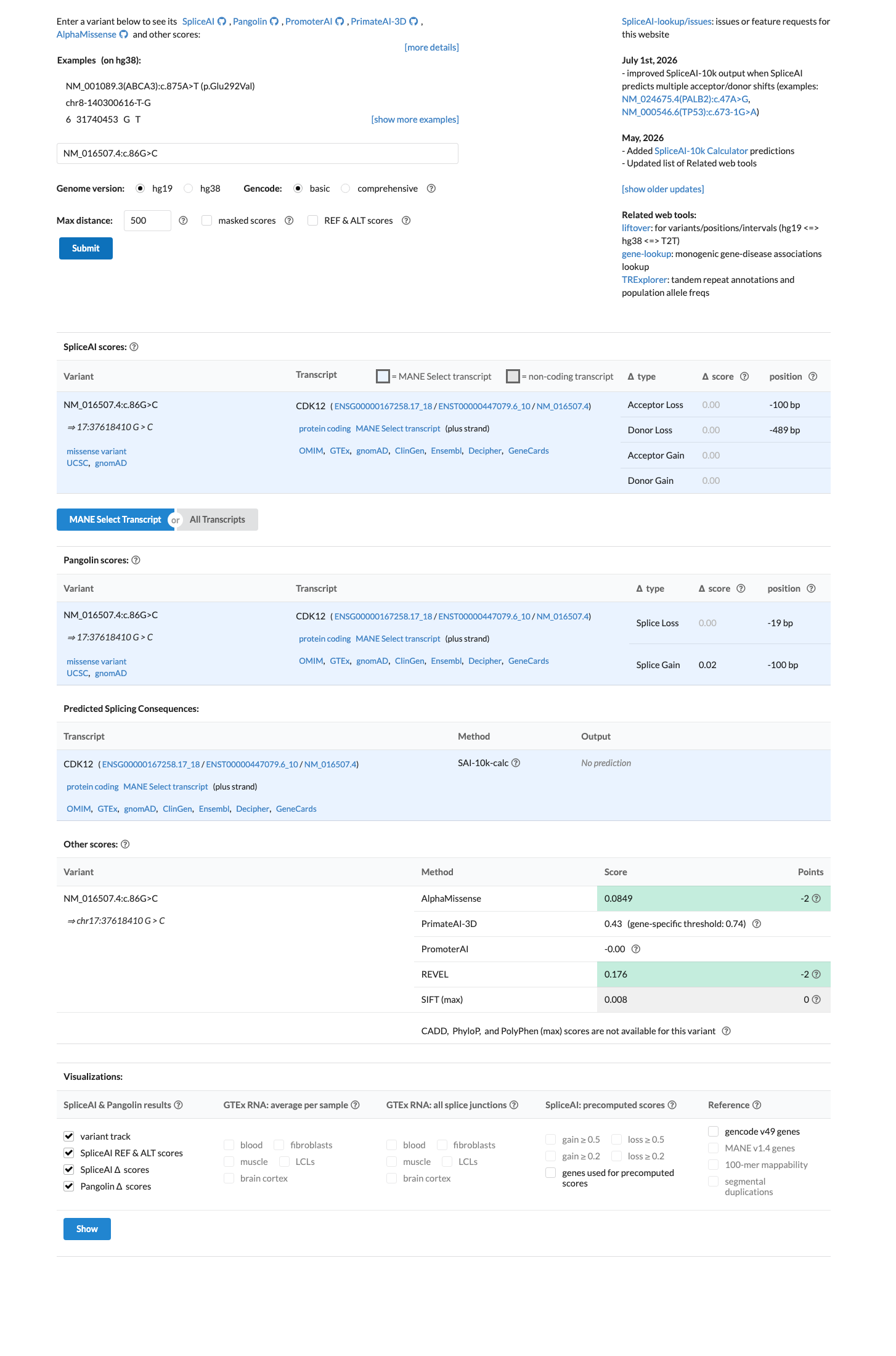
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.176. BayesDel score = -0.320226.
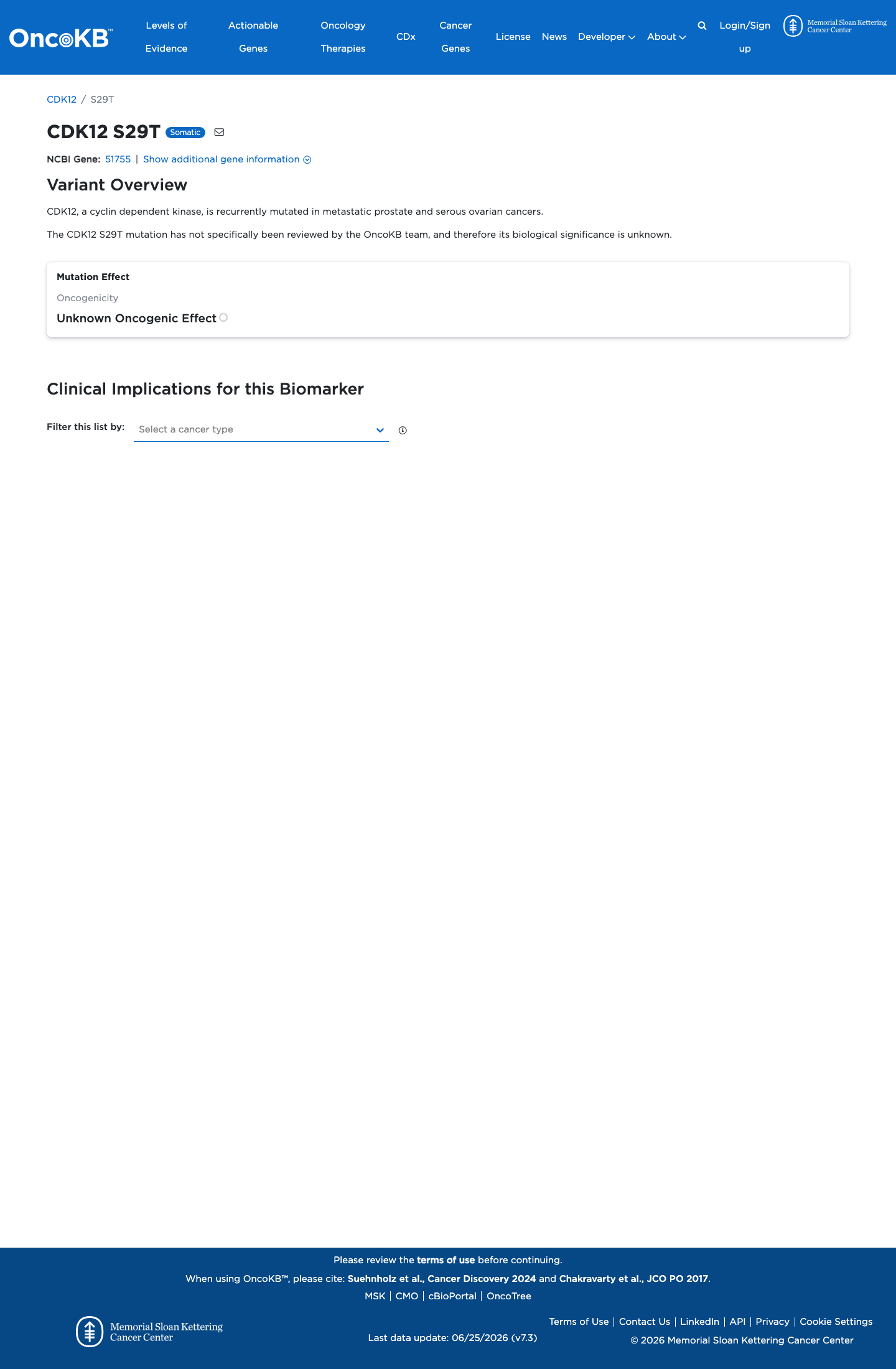
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. CDK12, a cyclin dependent kinase, is recurrently mutated in metastatic prostate and serous ovarian cancers.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links