NM_017745.5:c.519C>T is a synonymous variant (p.Ser173=) in exon 4 of BCOR that does not alter the protein sequence. SpliceAI predicts no impact on splicing (max delta score 0.00), consistent with BP7 (Supporting Benign).1 Three clinical laboratories in ClinVar classify this variant as Likely Benign, consistent with BP6 (Supporting Benign).2 The variant is observed in gnomAD at 0.035% in v4.1 (424 alleles) with no homozygotes, which is below the PM2 threshold of 0.1% but also below the BS1 threshold of 0.3%; population frequency is uninformative for this synonymous variant.3 No pathogenic or likely pathogenic ClinVar submissions, no functional evidence of damaging effect, and no segregation, de novo, or case-control data were identified to support a pathogenic classification.4
BCOR
Final classification
Likely Benign
BCOR c.519C>T · p.Ser173=
BCOR
NM_017745.5:c.519C>T is a synonymous variant (p.Ser173=) in exon 4 of BCOR that does not alter the protein sequence.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BP6 supporting benign, BP7 supporting benign; combination = 2 supporting benign, which maps to Likely Benign.
Classification rationale
BP6BP7
Likely Benign
BCOR c.519C>T
BP6 + BP7
→
Likely Benign
Gene diagram
· NM_017745.5 · variants mapped to exon structure
BCOR
NM_017745.5
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 19 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
BP6
supporting
Benign
Three independent clinical laboratories classify this variant as Likely Benign in ClinVar (Labcorp/Invitae SCV002392005, PreventionGenetics SCV004781451, CeGaT SCV004164852). While these are single-submitter assertions without expert panel review, the consistent direction across multiple clinical testing laboratories provides supporting evidence for a benign assessment.
Labcorp/Invitae (SCV002392005): Likely Benigncriteria providedPreventionGenetics (SCV004781451): Likely Benign
✓
BP7
supporting
Benign
Synonymous variant (p.Ser173=) with SpliceAI delta score of 0.00 across all categories (donor gain 0.00, donor loss 0.00, acceptor gain 0.00, acceptor loss 0.00), predicting no impact on splicing. The variant does not affect the splice consensus sequence nor create a novel splice site.
Synonymous substitution c.519C>T → p.(Ser173=)SpliceAI max delta 0.00 — no predicted donor/acceptor gain or lossNo alteration to canonical splice site motifs
Assessed · not applied
Pathogenic
PS2
No de novo evidence identified in ClinVar submissions, literature review, or population data.
PS3
No functional studies demonstrating a damaging effect on the gene or gene product have been identified.
PS4
No case-control enrichment data or statistically significant excess of variant in affected individuals versus controls.
PM1
This synonymous variant is located in exon 4 of BCOR (c.519).
PM2
The variant is present in gnomAD with 34 alleles in v2.1 (AF 0.017%) and 424 alleles in v4.1 (AF 0.035%).
PM6
No de novo occurrence (with maternity and paternity confirmed) has been reported for this variant in the reviewed literature or ClinVar submissions.
PP1
No cosegregation data available.
PP2
BCOR is not established as a gene with a low rate of benign missense variation where missense changes are a common mechanism of disease.
PP3
No in silico evidence supports a pathogenic effect.
PP4
No patient phenotype data specific to a BCOR-related disorder is available for review.
PP5
No reputable source classifies this variant as pathogenic.
Benign
BA1
Maximum population allele frequency is 0.043% (NFE in gnomAD v4.1), well below the 1% BA1 threshold.
BS1
Maximum population allele frequency is 0.043% (NFE in gnomAD v4.1), below the 0.3% BS1 threshold for non-VCEP assessment.
BS2
No homozygous observations in gnomAD (0 homozygotes across v2.1 and v4.1).
BS3
No functional studies demonstrating no deleterious effect have been identified.
BS4
No segregation data demonstrating lack of cosegregation with disease in affected family members.
BP2
No evidence of this variant observed in trans with a known pathogenic BCOR variant.
BP4
Insufficient multiple lines of computational evidence to classify as benign.
BP5
No case reports identifying this variant in an individual with an alternative molecular basis for disease have been identified.
N/A · 4
PVS1 · PS1 · PM5 · BP1
Research & evidence
Population frequency · supports benign
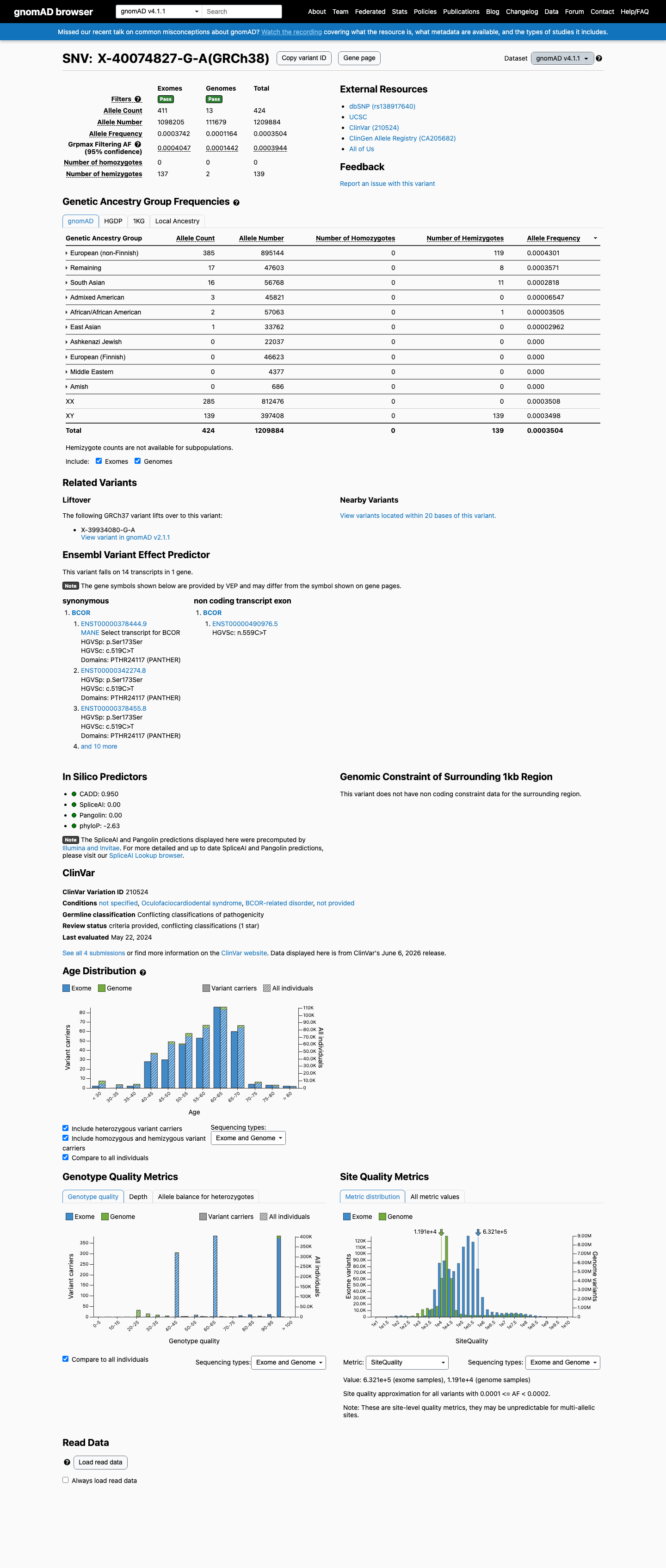
gnomAD v4.1
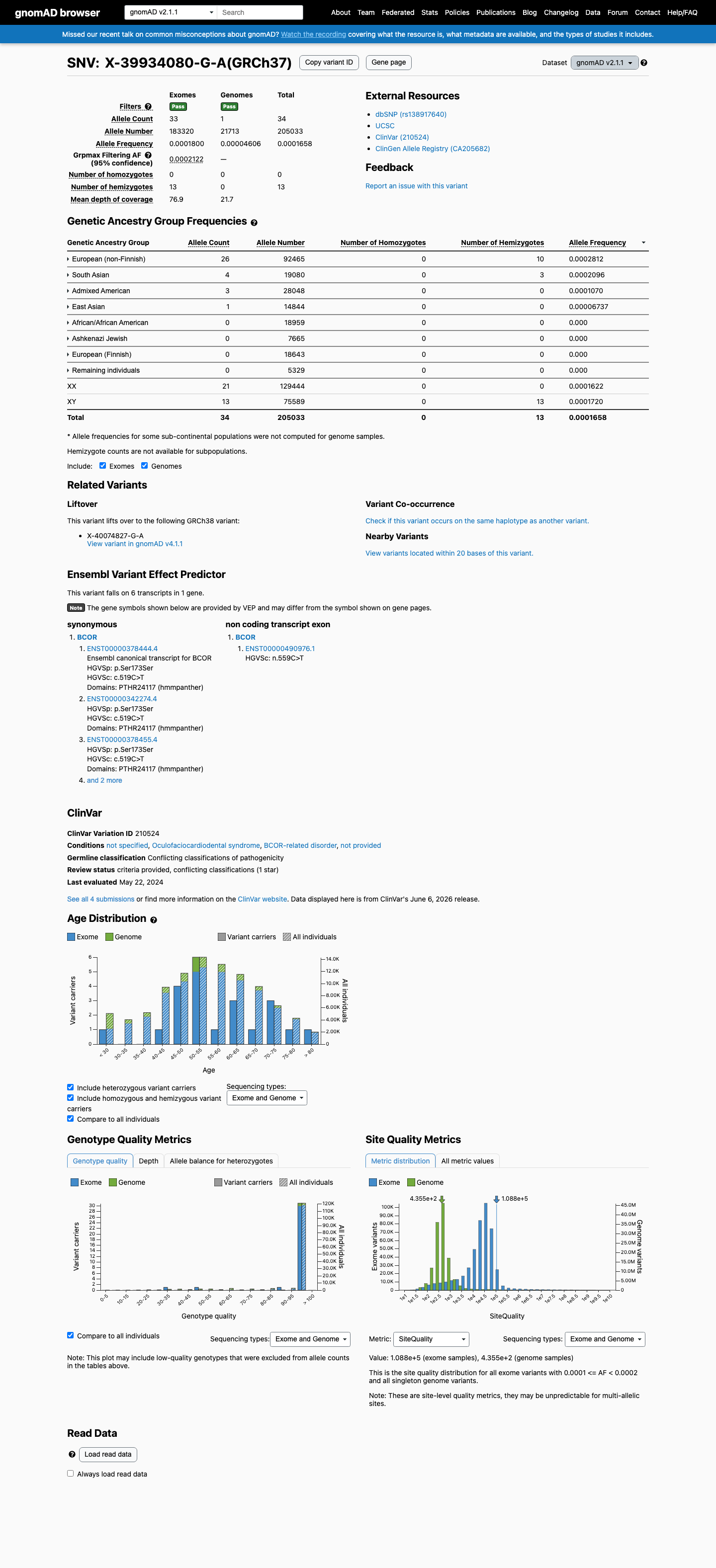
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000350447; MAF= 0.03504%, 424/1209884 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 0.000430098; MAF= 0.04301%, 385/895144 alleles, homozygotes = 0); grpmax FAF= 0.00039444.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.000165827; MAF= 0.01658%, 34/205033 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 0.000281187; MAF= 0.02812%, 26/92465 alleles, homozygotes = 0); grpmax FAF= 0.00021222.
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.035%
· 424 / 1,209,884
0 hom · FAF 0.039%
0 hom · FAF 0.039%
European (non-Finnish) 385 / 895,144 |
0.043% |
Remaining individuals 17 / 47,603 |
0.036% |
South Asian 16 / 56,768 |
0.028% |
Admixed American 3 / 45,821 |
0.0065% |
African/African American 2 / 57,063 |
0.0035% |
East Asian 1 / 33,762 |
0.003% |
+ 4 not observed (European (Finnish), Amish, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.017%
· 34 / 205,033
0 hom · FAF 0.021%
0 hom · FAF 0.021%
European (non-Finnish) 26 / 92,465 |
0.028% |
South Asian 4 / 19,080 |
0.021% |
Admixed American 3 / 28,048 |
0.011% |
East Asian 1 / 14,844 |
0.0067% |
+ 4 not observed (African/African American, Ashkenazi Jewish, European (Finnish), Remaining individuals)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
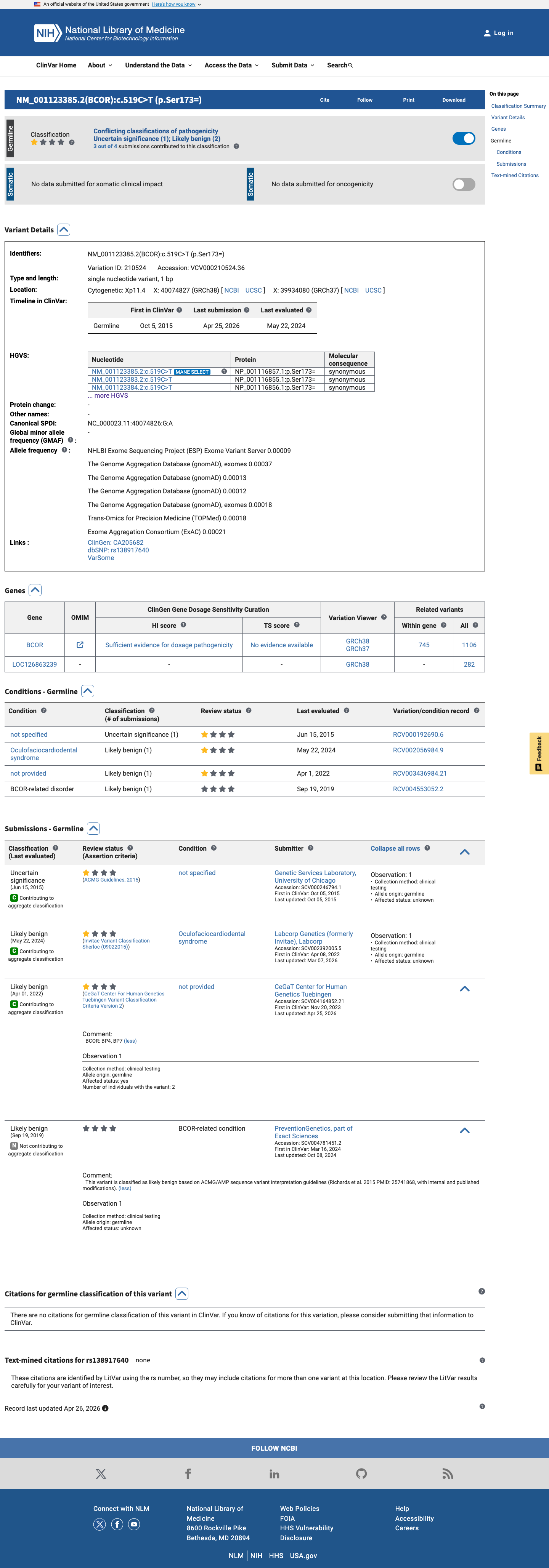
ClinVar
This variant is present in ClinVar (Variation ID: 210524); submission details unavailable.
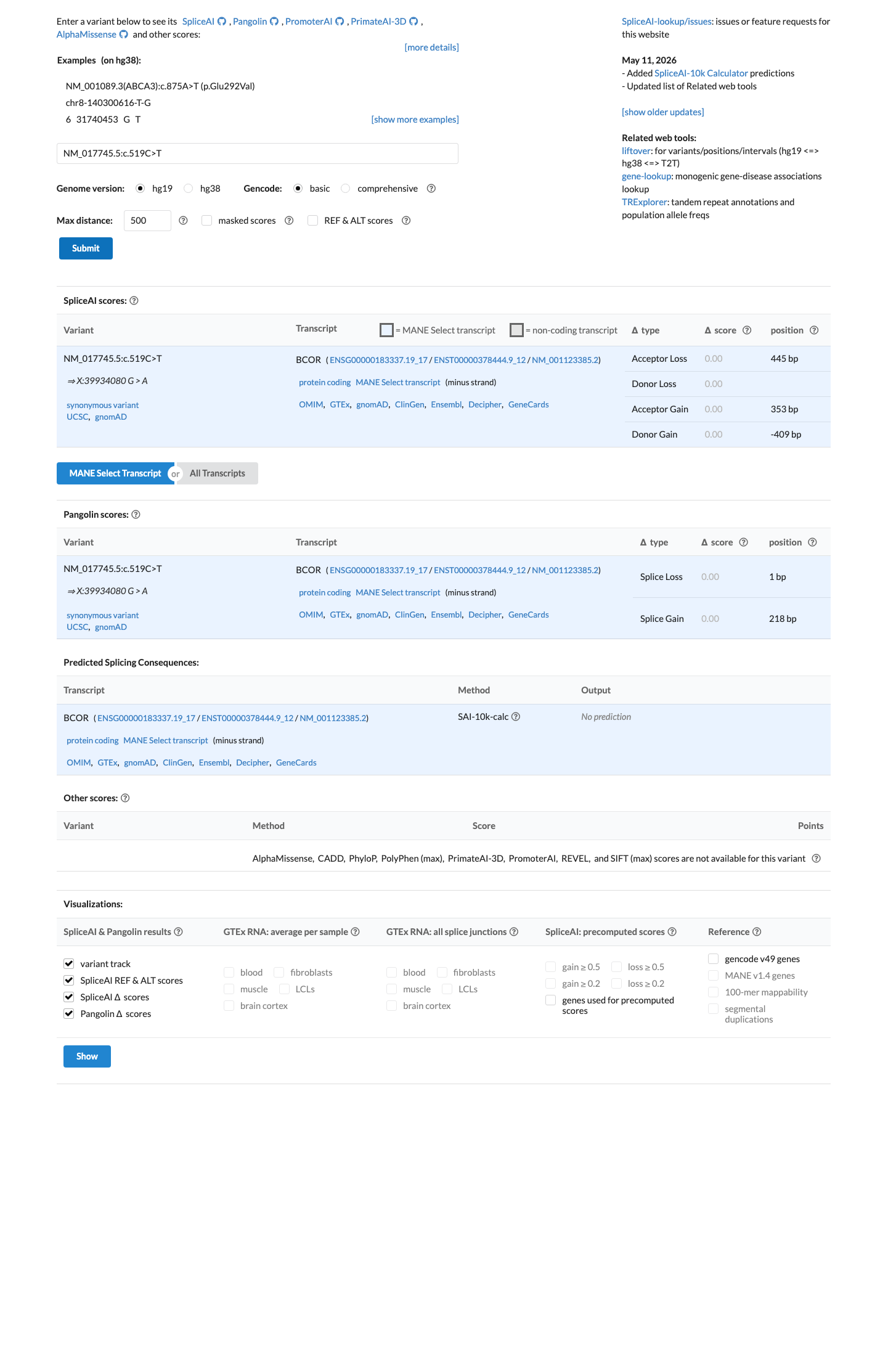
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV60708452, n = 1 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 2 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR