PVS1 (very strong): NM_022552.4:c.1154delC is a frameshift deletion introducing a premature termination codon at position 406 (NP_072046.2:p.Pro385ArgfsTer22) in exon 10 of 22 coding exons, predicted to trigger nonsense-mediated decay. DNMT3A loss of function is an established disease mechanism for Tatton-Brown-Rahman syndrome, with a ClinGen haploinsufficiency score of 3.1 PM2 (moderate): The variant is absent from gnomAD v2.1 (0/250,600 alleles) and present at extremely low frequency in gnomAD v4.1 (6/1,613,666 alleles; AF=3.72e-06). The highest subpopulation frequency is 1.67e-05 in the Admixed American population, well below the 0.1% PM2 threshold.2 Combined classification: One very strong criterion (PVS1) and one moderate criterion (PM2) satisfy the Likely Pathogenic threshold under generic ACMG/AMP 2015 rules (1 Very Strong + 1 Moderate).3
DNMT3A
Final classification
Likely Pathogenic
DNMT3A c.1154del · p.Pro385ArgfsTer22
DNMT3A
PVS1 (very strong): NM_022552.4:c.1154delC is a frameshift deletion introducing a premature termination codon at position 406 (NP_072046.2:p.Pro385ArgfsTer22) in exon 10 of 22 coding exons, predicted to trigger nonsense-mediated decay. DNMT3A loss of function is an established disease mechanism for Tatton-Brown-Rahman syndrome, with a ClinGen haploinsufficiency score of 3.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 very strong, PM2 moderate; combination = 1 very strong + 1 moderate, which maps to Likely Pathogenic.
Classification rationale
PVS1PM2
Likely Pathogenic
DNMT3A c.1154del
PVS1 + PM2
→
Likely Pathogenic
1
pvs1_generic_framework ↗pvs1_gene_contextpvs1_variant_assessment
3
generic_acmg_combination_rules
Gene diagram
· NM_022552.4 · variants mapped to exon structure
DNMT3A
NM_022552.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 16 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
NM_022552.4:c.1154delC is a frameshift deletion predicted to produce a premature termination codon at residue 406 (NP_072046.2:p.Pro385ArgfsTer22) in exon 10 of 22 coding exons. The variant is predicted to trigger nonsense-mediated decay. DNMT3A loss of function is an established disease mechanism for Tatton-Brown-Rahman syndrome (ClinGen haploinsufficiency score=3). Under ClinGen SVI PVS1 recommendations (PMC6185798), a null variant in a gene where LOF is a known mechanism qualifies for PVS1 at very strong level.
Frameshift deletion introducing PTC at codon 406 in exon 10/22predicted to trigger NMDDNMT3A LOF is an established mechanism for Tatton-Brown-Rahman syndrome (ClinGen haploinsufficiency score=3)
✓
PM2
moderate
Pathogenic
NM_022552.4:c.1154delC is absent from gnomAD v2.1 (0/250,600 alleles) and present at extremely low frequency in gnomAD v4.1 (6/1,613,666 alleles, AF=3.72e-06). Complete absence from the v2.1 dataset and near-absence from v4.1 satisfies PM2 under generic ACMG guidelines at moderate strength.
gnomAD v2.1: 0/250600 alleles (completely absent)gnomAD v4.1: 6/1
Assessed · not applied
Pathogenic
PS2
PS2 requires a de novo occurrence with confirmed paternity and maternity.
PS3
PS3 requires well-established in vitro or in vivo functional studies demonstrating a damaging effect specific to this variant.
PS4
PS4 requires a statistically significant enrichment of the variant in affected individuals compared with controls.
PM1
PM1 requires location in a mutational hot spot or critical functional domain without benign variation.
PM6
PM6 requires an assumed de novo occurrence without confirmation of paternity and maternity.
PP1
PP1 requires cosegregation of the variant with disease in multiple affected family members.
PP4
PP4 requires that the patient's phenotype or family history is highly specific for a disease with a single genetic etiology.
PP5
PP5 requires that a reputable source recently reports the variant as pathogenic but the evidence is not independently verifiable.
Benign
BA1
BA1 requires an allele frequency >1% in any general population.
BS1
BS1 requires an allele frequency greater than expected for the disorder.
BS2
BS2 requires observation of the variant in a healthy adult individual for a disorder with full penetrance expected at an early age.
BS3
BS3 requires well-established in vitro or in vivo functional studies showing no damaging effect on protein function or splicing.
BS4
BS4 requires lack of segregation of the variant with disease in affected family members.
BP2
BP2 requires observation of the variant in trans with a known pathogenic variant in a fully penetrant dominant disorder, or in cis with a pathogenic variant in any inheritance pattern.
BP5
BP5 requires that the variant is found in a case with an alternate molecular basis for disease.
BP6
BP6 requires that a reputable source reports the variant as benign but the evidence is not independently verifiable.
N/A · 9
PS1 · PM4 · PM5 · PP2 · PP3 · BP1 · BP3 · BP4 · BP7
Research & evidence
Population frequency · supports pathogenic
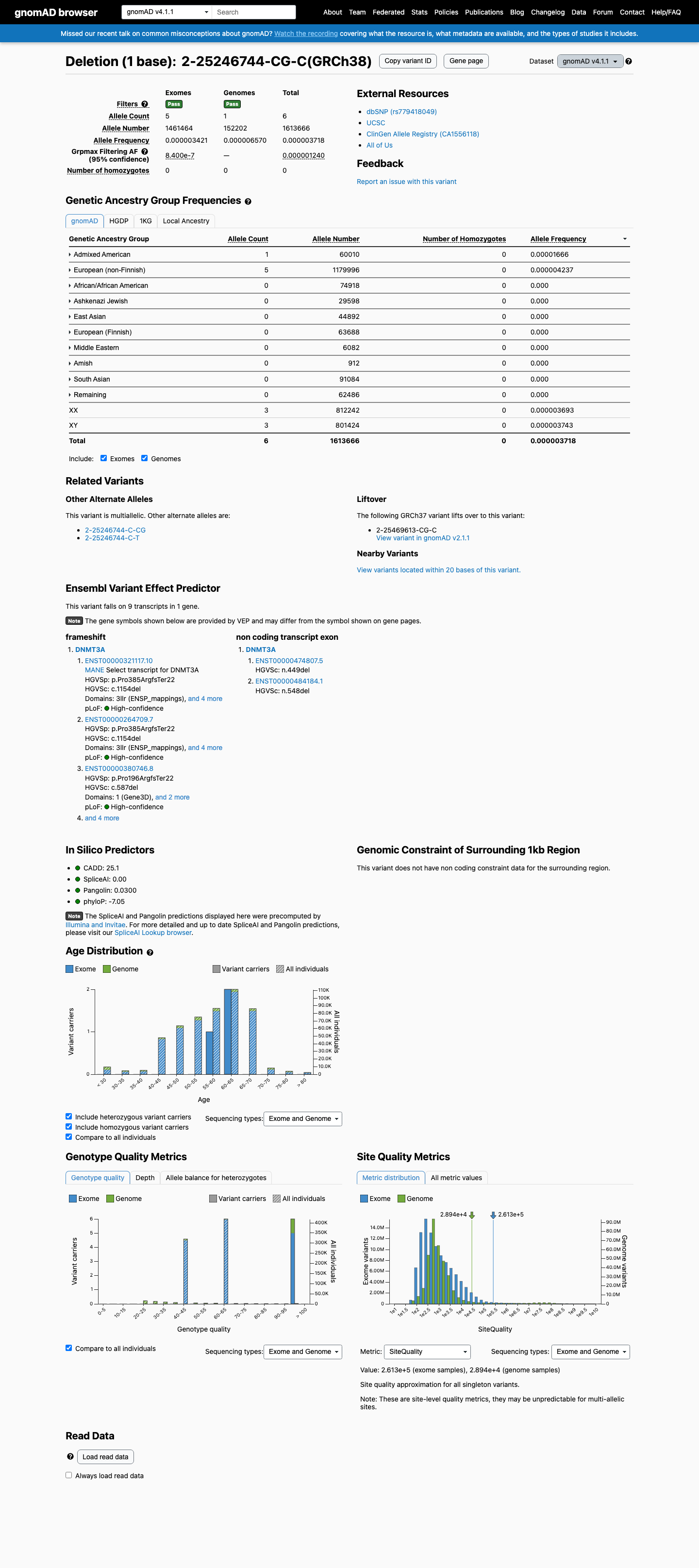
gnomAD v4.1
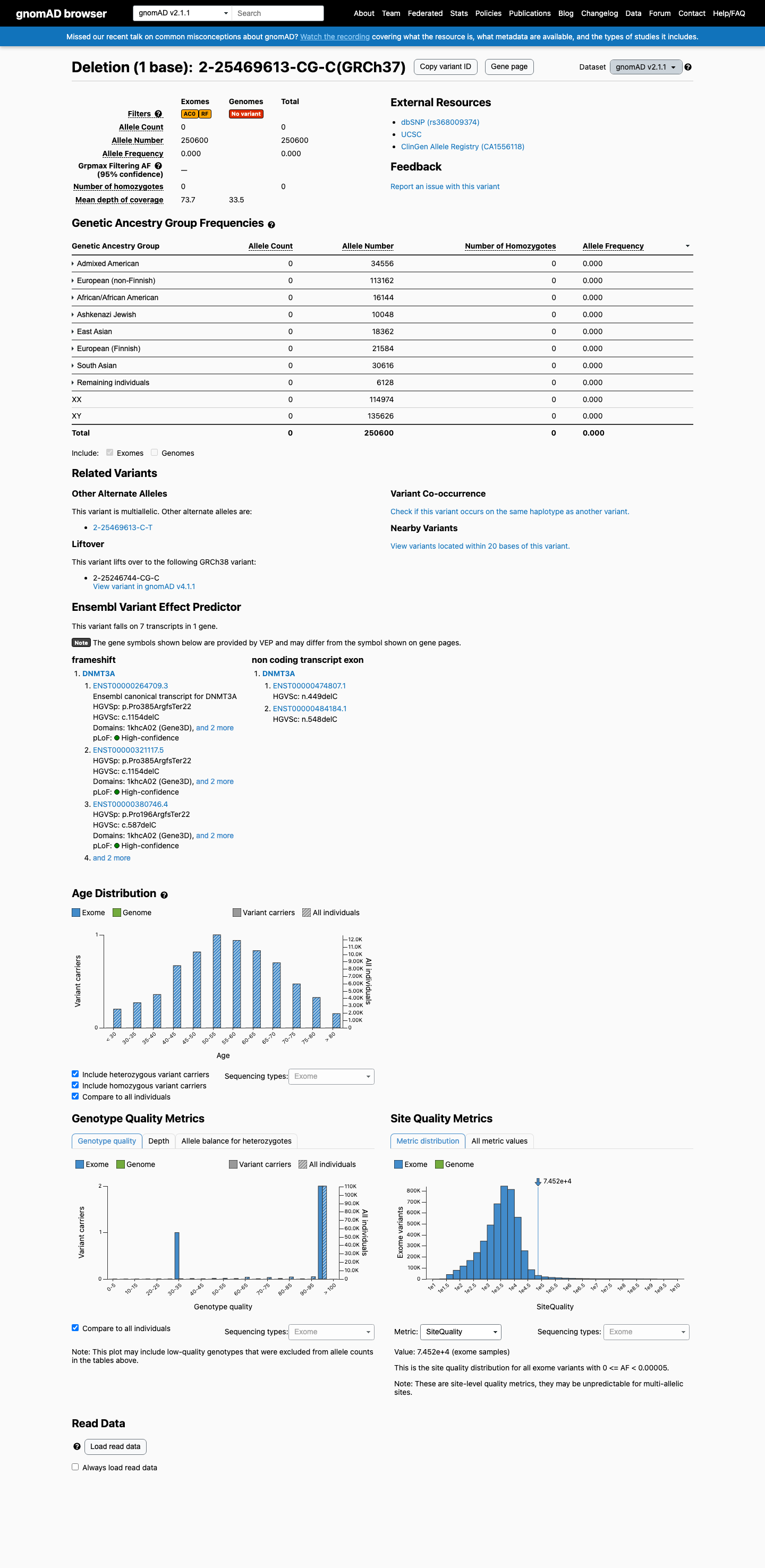
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 3.71824e-06; MAF= 0.00037%, 6/1613666 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 1.66639e-05; MAF= 0.00167%, 1/60010 alleles, homozygotes = 0); grpmax FAF= 1.24e-06.
v2.1
This variant is present in gnomAD v2.1 (AF= 0; MAF= 0.00000%, 0/250600 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0; MAF= 0.00000%, 0/16144 alleles, homozygotes = 0).
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00037%
· 6 / 1,613,666
0 hom · FAF 0.00012%
0 hom · FAF 0.00012%
Admixed American 1 / 60,010 |
0.0017% |
European (non-Finnish) 5 / 1,179,996 |
0.00042% |
+ 8 not observed (Remaining individuals, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / 250,600
0 hom
0 hom
Not observed in any ancestry group.
+ 8 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
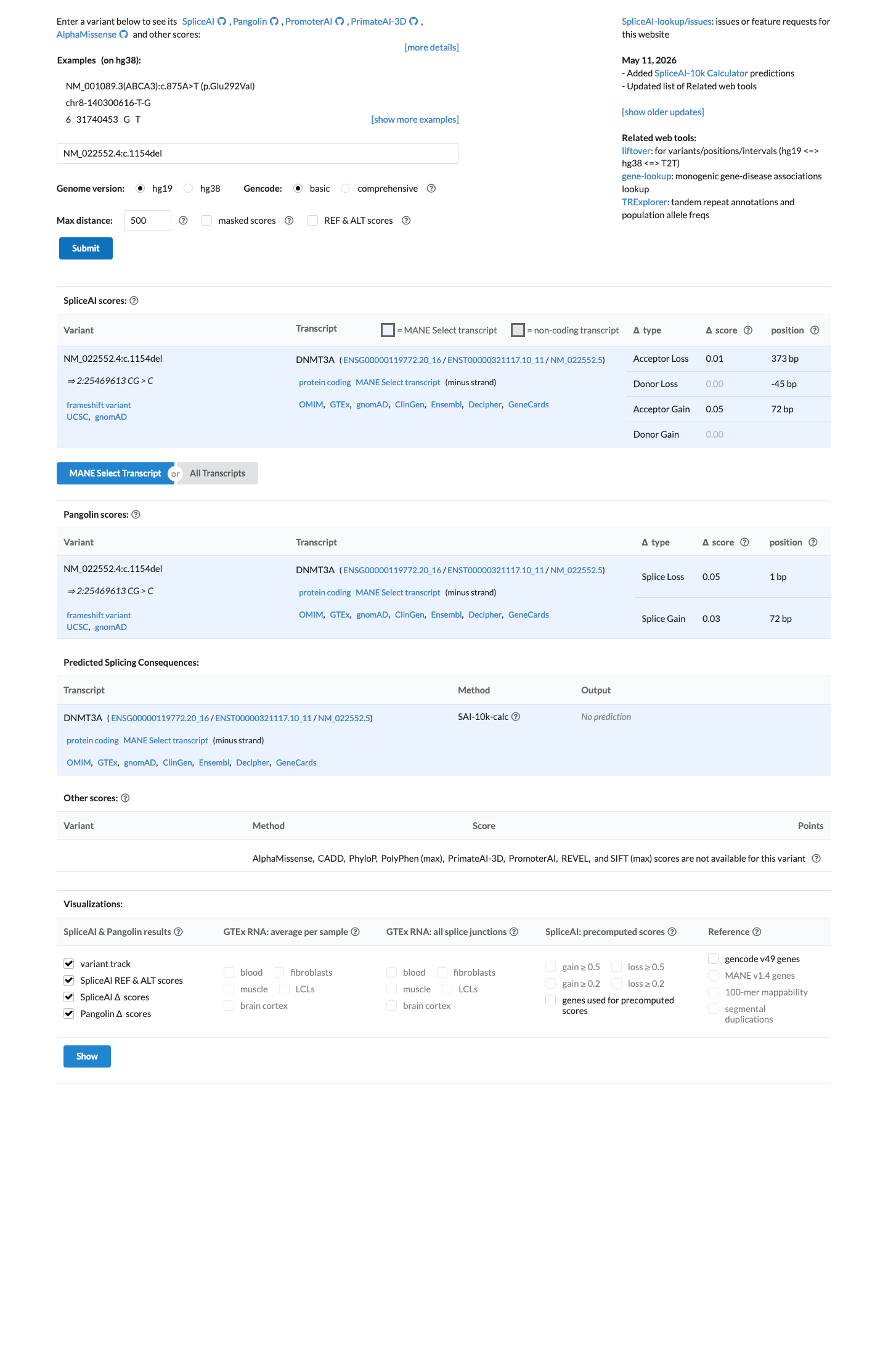
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.05).
Functional
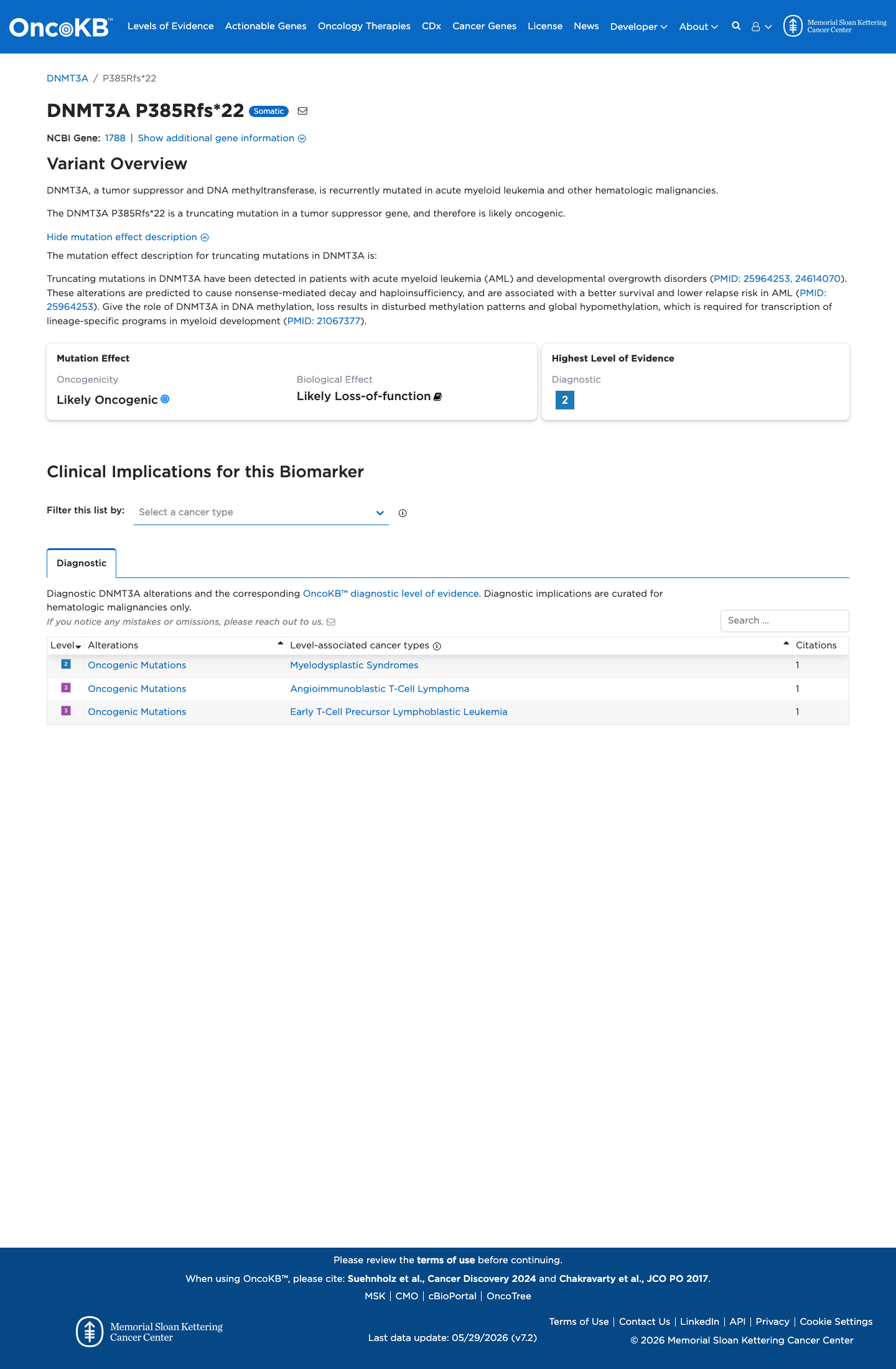
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
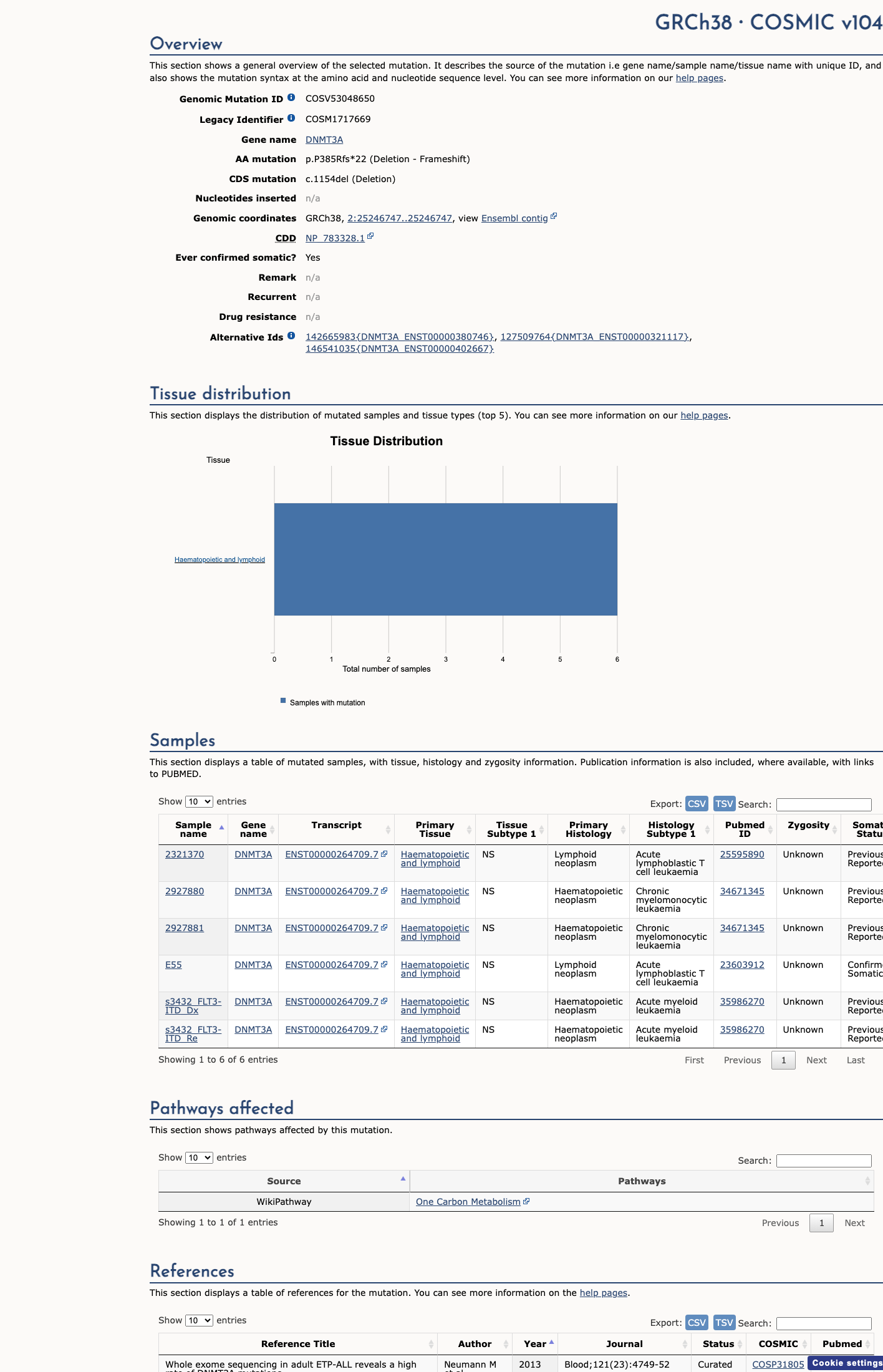
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV53048650, n = 6 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 3 PMIDs not cited in assessment
24614070 ↗
Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability.
ONCOKB
25964253 ↗
Simpson's Paradox and the Impact of Different DNMT3A Mutations on Outcome in Younger Adults With Acute Myeloid Leukemia.
ONCOKB