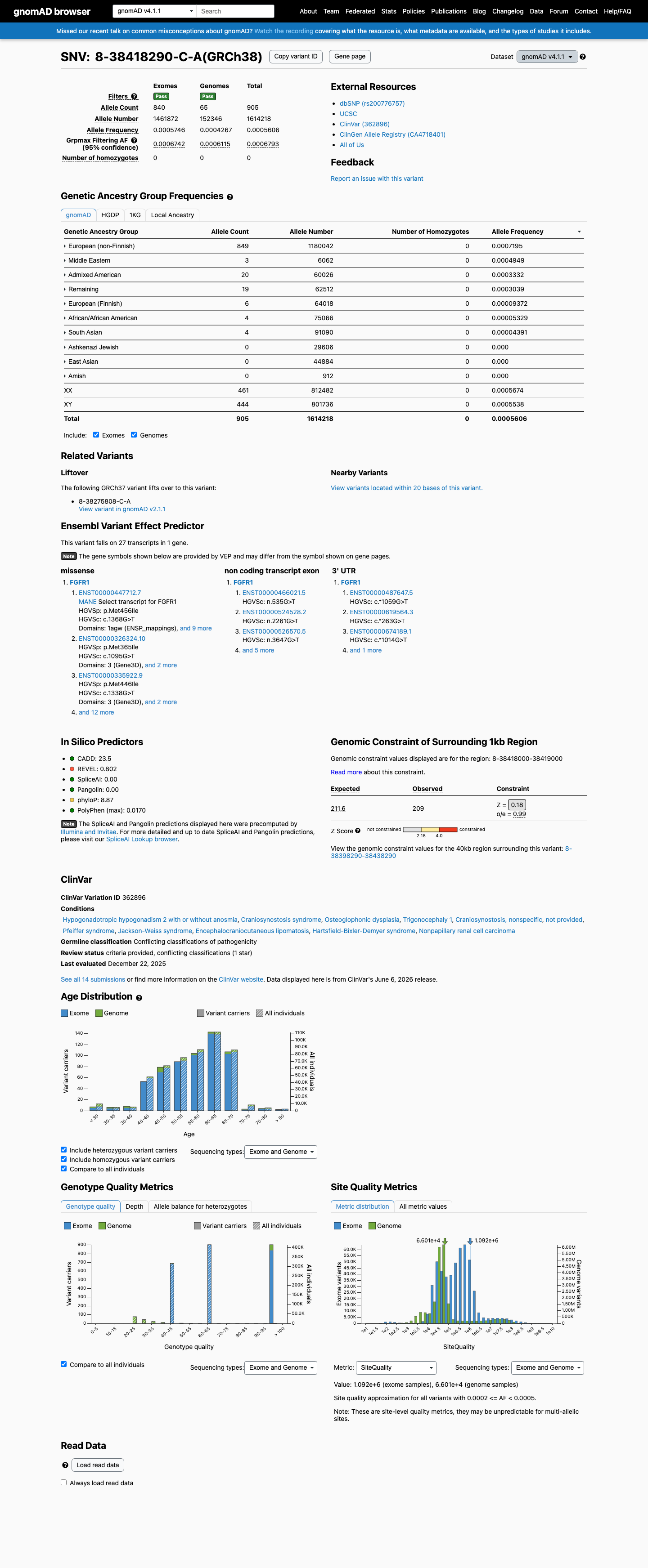
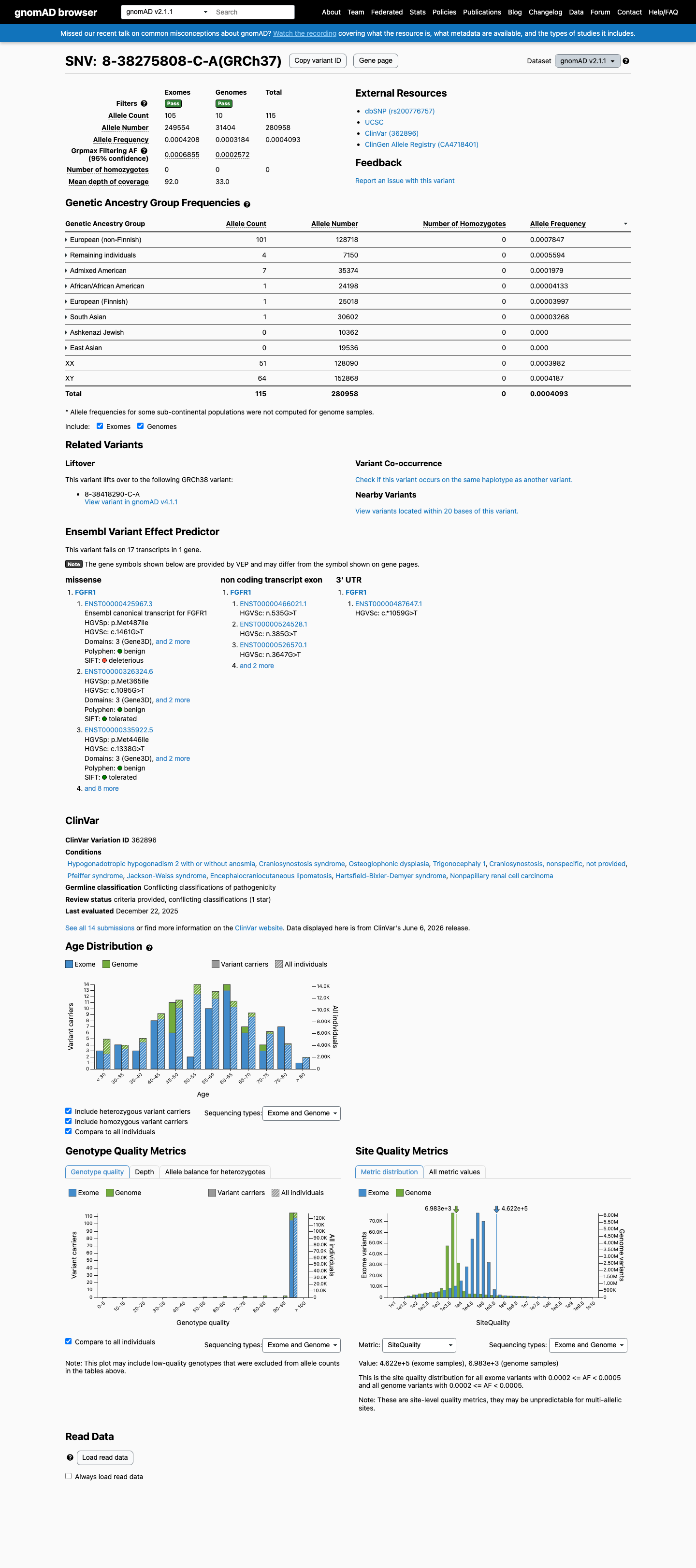
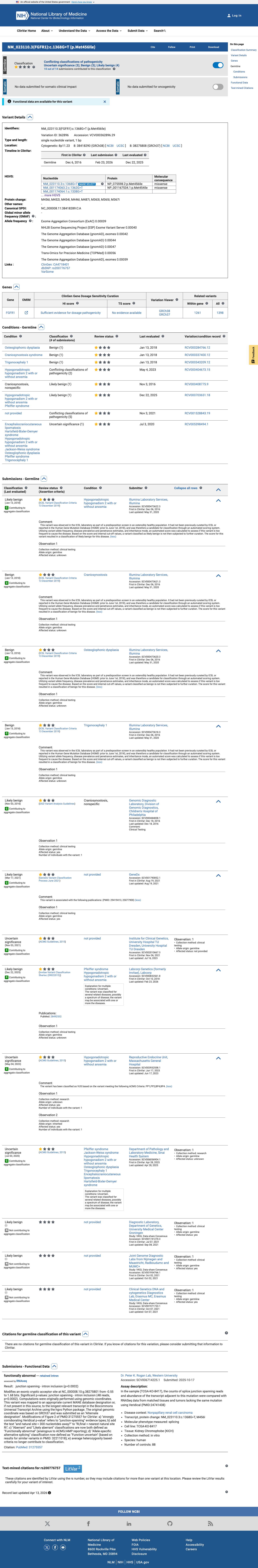
NM_023110.3:c.1368G>T (p.Met456Ile) is a missense variant in exon 10 of FGFR1, encoding the tyrosine kinase domain. This variant is present in gnomAD v2.1 at an allele frequency of 0.041% (115/280,958 alleles, 0 homozygotes) and in v4.1 at 0.056% (905/1,614,218 alleles, 0 homozygotes), with a grpmax filtering allele frequency of 0.069%. While below the formal BS1 threshold of 0.3%, the substantial carrier count in population databases (905 in v4.1) indicates this variant is not a rare private mutation.1 In ClinVar, 10 of 11 clinical laboratory submissions classify this variant as Likely benign (7) or Benign (3), with one VUS. No submitter classifies the variant as pathogenic or likely pathogenic. The review status is 'criteria provided, single submitter' without expert panel consensus.2 In silico predictions are discordant: REVEL (0.802) predicts damaging, while BayesDel (0.069) and SpliceAI (max delta 0.00) predict benign/no splicing impact. No variant-specific functional studies or case-level observations of pathogenicity were identified in the reviewed literature.3 Applying generic ACMG/AMP 2015 combination rules: one supporting benign criterion (BP6) is met. PM2 is met at supporting level for pathogenicity but is substantially weakened by the high carrier count (905 in v4.1). The overall evidence profile favors a likely benign interpretation based on population frequency and multi-laboratory clinical consensus, though the formal BS1 threshold is not crossed.4
FGFR1
Final classification
VUS
FGFR1 c.1368G>T · p.Met456Ile
FGFR1
NM_023110.3:c.1368G>T (p.Met456Ile) is a missense variant in exon 10 of FGFR1, encoding the tyrosine kinase domain.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP6 supporting; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP6
VUS
FGFR1 c.1368G>T
PM2 + BP6
→
VUS
Gene diagram
· NM_023110.3 · variants mapped to exon structure
FGFR1
NM_023110.3
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 21 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
review
Pathogenic
The variant is present in gnomAD at low frequency — v2.1 AF 0.041% (115/280,958 alleles) and v4.1 AF 0.056% (905/1,614,218 alleles), both below the 0.1% threshold for PM2. However, the substantial carrier count (905 in v4.1) substantially weakens the evidence weight and this criterion should be considered borderline in the context of a rare disease gene.
gnomAD v2.1 AF=0.041% (115/280958)v4.1 AF=0.056% (905/1
✓
BP6
supporting
Benign
Ten of eleven clinical laboratory submissions in ClinVar classify NM_023110.3:c.1368G>T as Likely benign (7 labs) or Benign (3 labs), with only one VUS. While no expert panel review is available, the strong inter-laboratory consensus towards benign supports BP6 at the supporting level.
ClinVar: 7 Likely benign3 Benign1 VUS. Majority consensus supports benign classification.
Assessed · not applied
Pathogenic
PS1
No ClinVar or literature evidence identifies a different nucleotide change at the same codon (c.1368) producing the same amino acid change (p.Met456Ile).
PS2
No de novo observation data are available for this variant.
PS3
No well-established in vitro or in vivo functional studies have been identified for p.Met456Ile.
PS4
No case-control study or systematic phenotypic data are available to demonstrate significantly increased prevalence of this variant in affected individuals versus controls.
PM1
Although p.Met456Ile resides in the tyrosine kinase domain of FGFR1, the variant is not located in a statistically significant mutational hotspot per the hotspot screen.
PM5
Automated PM5 candidate harvesting found no same-residue (Met456) comparator variants with pathogenic or likely pathogenic classification in ClinVar.
PM6
No de novo observation with unconfirmed parentage has been reported for this variant.
PP1
No family segregation data are available for NM_023110.3:c.1368G>T.
PP2
FGFR1 has a mix of pathogenic and benign missense variants; it does not exhibit a low rate of benign missense variation that would support PP2.
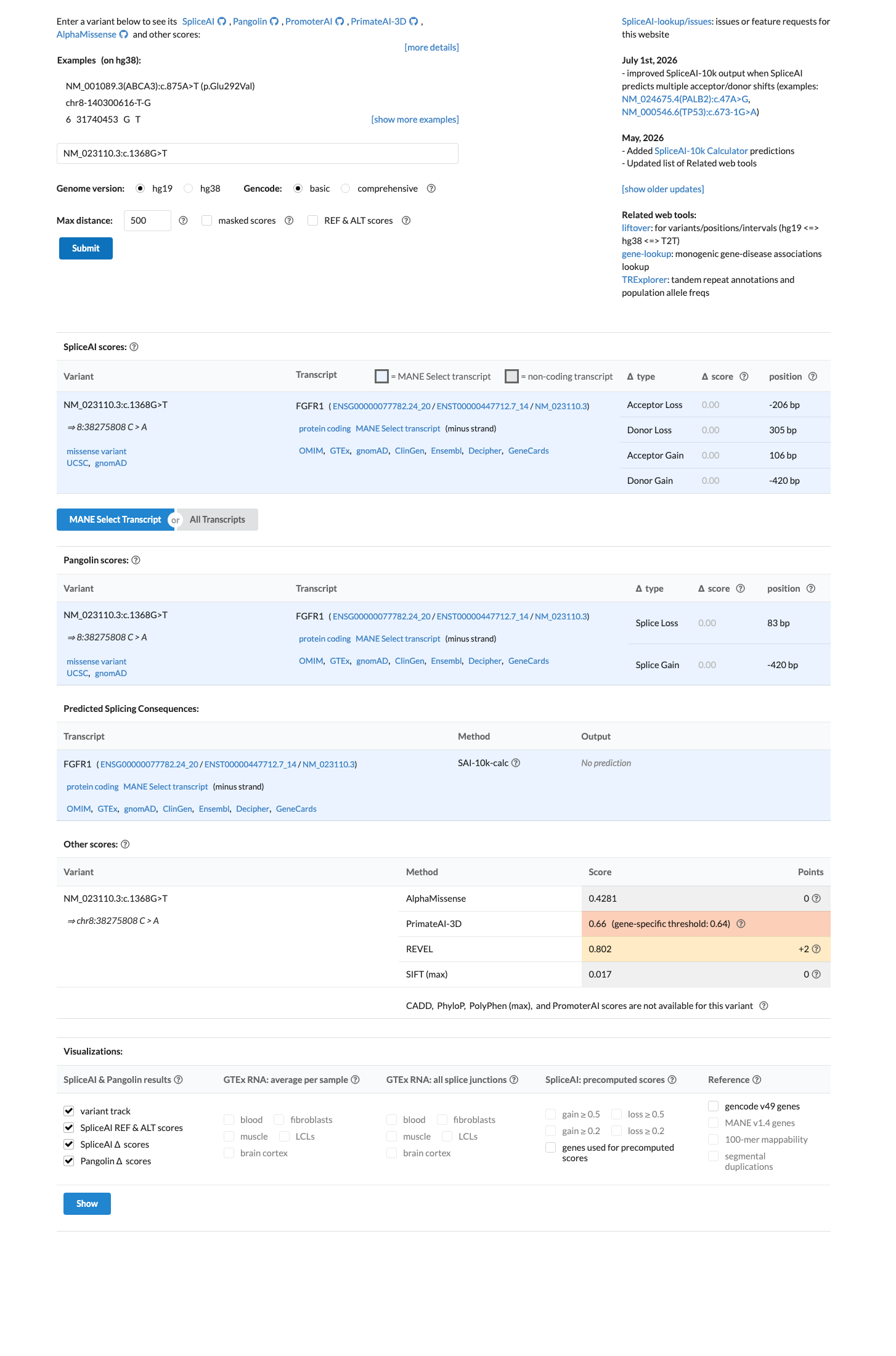
PP3
In silico predictions are discordant: REVEL score 0.802 suggests a damaging effect, but BayesDel score 0.069 predicts benign and SpliceAI delta 0.00 predicts no splicing impact.
PP4
No patient phenotype information is available in the case data.
PP5
ClinVar reports a consensus classification of Likely benign/Benign from 10 of 11 clinical laboratories, with only 1 VUS.
Benign
BA1
The maximum allele frequency in gnomAD is 0.056% (v4.1) with grpmax FAF of 0.069%, well below the 1% threshold required for BA1.
BS1
The maximum allele frequency in gnomAD is 0.056% (v4.1) with grpmax FAF of 0.069%, below the 0.3% threshold for BS1.
BS2
No homozygous individuals are observed in gnomAD (v2.1 or v4.1).
BS3
No well-established in vitro or in vivo functional studies demonstrate that p.Met456Ile has no deleterious effect.
BS4
No family segregation data are available to demonstrate lack of cosegregation with disease in affected family members.
BP1
FGFR1-related disorders are not solely caused by loss-of-function truncating variants; activating missense variants in the tyrosine kinase domain are a well-established pathogenic mechanism (e.g., craniosynostosis, Hartsfield syndrome, osteoglophonic dysplasia).
BP2
No data are available regarding observation of this variant in trans with a known pathogenic dominant FGFR1 variant.
BP4
In silico predictions are discordant: REVEL 0.802 predicts damaging, while BayesDel 0.069 and SpliceAI 0.00 predict benign/neutral.
BP5
No data are available indicating that this variant has been observed in a case with an alternative molecular cause for disease.
N/A · 5
PVS1 · PM3 · PM4 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000560643; MAF= 0.05606%, 905/1614218 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 0.000719466; MAF= 0.07195%, 849/1180042 alleles, homozygotes = 0); grpmax FAF= 0.00067933.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.000409314; MAF= 0.04093%, 115/280958 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 0.000784661; MAF= 0.07847%, 101/128718 alleles, homozygotes = 0); grpmax FAF= 0.00068549.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.00043426338074041906, 8/18422 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.056%
· 905 / 1,614,218
0 hom · FAF 0.068%
0 hom · FAF 0.068%
European (non-Finnish) 849 / 1,180,042 |
0.072% |
Middle Eastern 3 / 6,062 |
0.049% |
Admixed American 20 / 60,026 |
0.033% |
Remaining individuals 19 / 62,512 |
0.03% |
European (Finnish) 6 / 64,018 |
0.0094% |
African/African American 4 / 75,066 |
0.0053% |
South Asian 4 / 91,090 |
0.0044% |
+ 3 not observed (Amish, East Asian, Ashkenazi Jewish)
gnomAD v2.1
0.041%
· 115 / 280,958
0 hom · FAF 0.069%
0 hom · FAF 0.069%
European (non-Finnish) 101 / 128,718 |
0.078% |
Remaining individuals 4 / 7,150 |
0.056% |
Admixed American 7 / 35,374 |
0.02% |
African/African American 1 / 24,198 |
0.0041% |
European (Finnish) 1 / 25,018 |
0.004% |
South Asian 1 / 30,602 |
0.0033% |
+ 2 not observed (Ashkenazi Jewish, East Asian)
gnomAD Canada 🇨🇦
0.043%
· 8 / 18,422
0 hom · FAF 0.034%
0 hom · FAF 0.034%
European (non-Finnish) 8 / 11,742 |
0.068% |
+ 8 not observed (African/African American, Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, Remaining individuals, South Asian)
ClinVar
This variant has been reported in ClinVar as Likely benign (7 clinical laboratories) and as Benign (3 clinical laboratories) and as Uncertain significance (1 clinical laboratory). (ClinVarID = 362896)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.802. BayesDel score = 0.0686773.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. FGFR1, a receptor tyrosine kinase, is altered by mutation, chromosomal rearrangement or amplification in various cancer types including lung and breas

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 7 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR