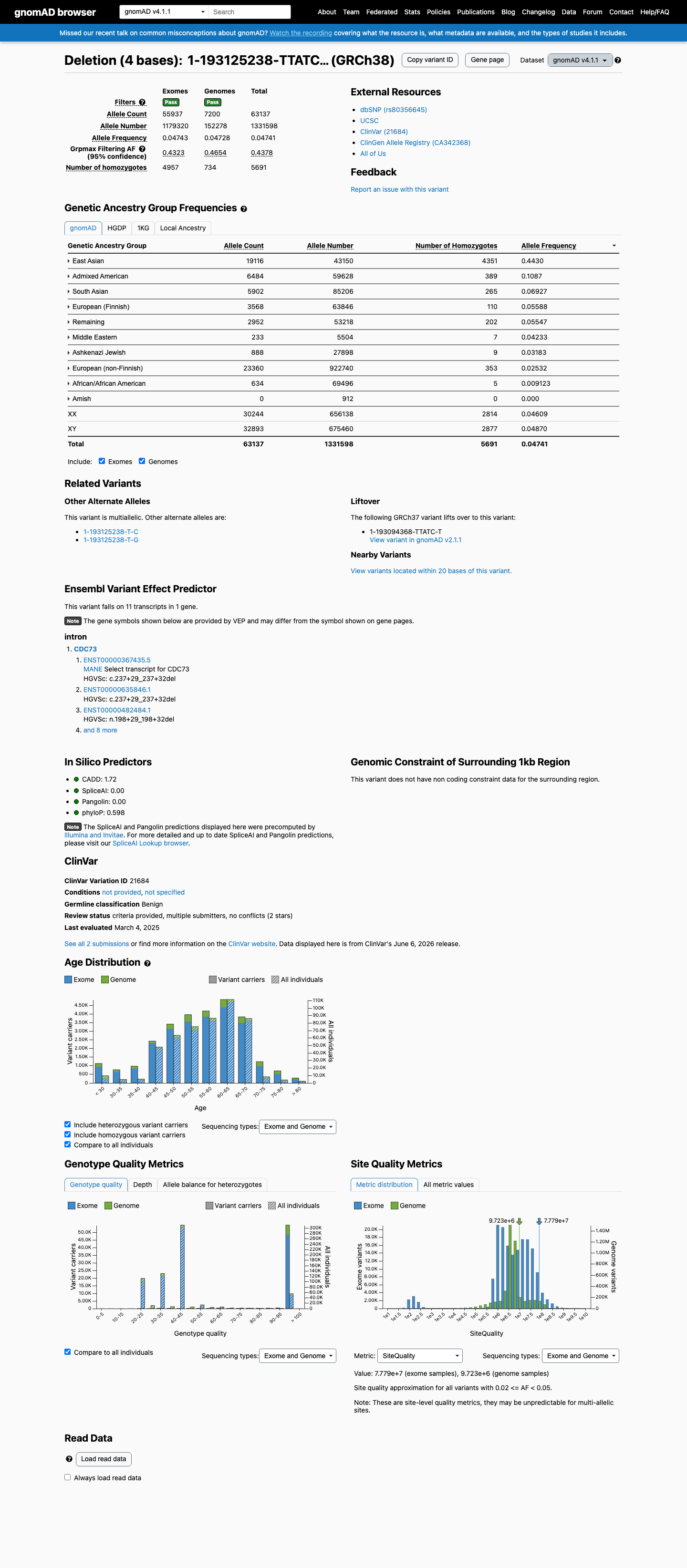
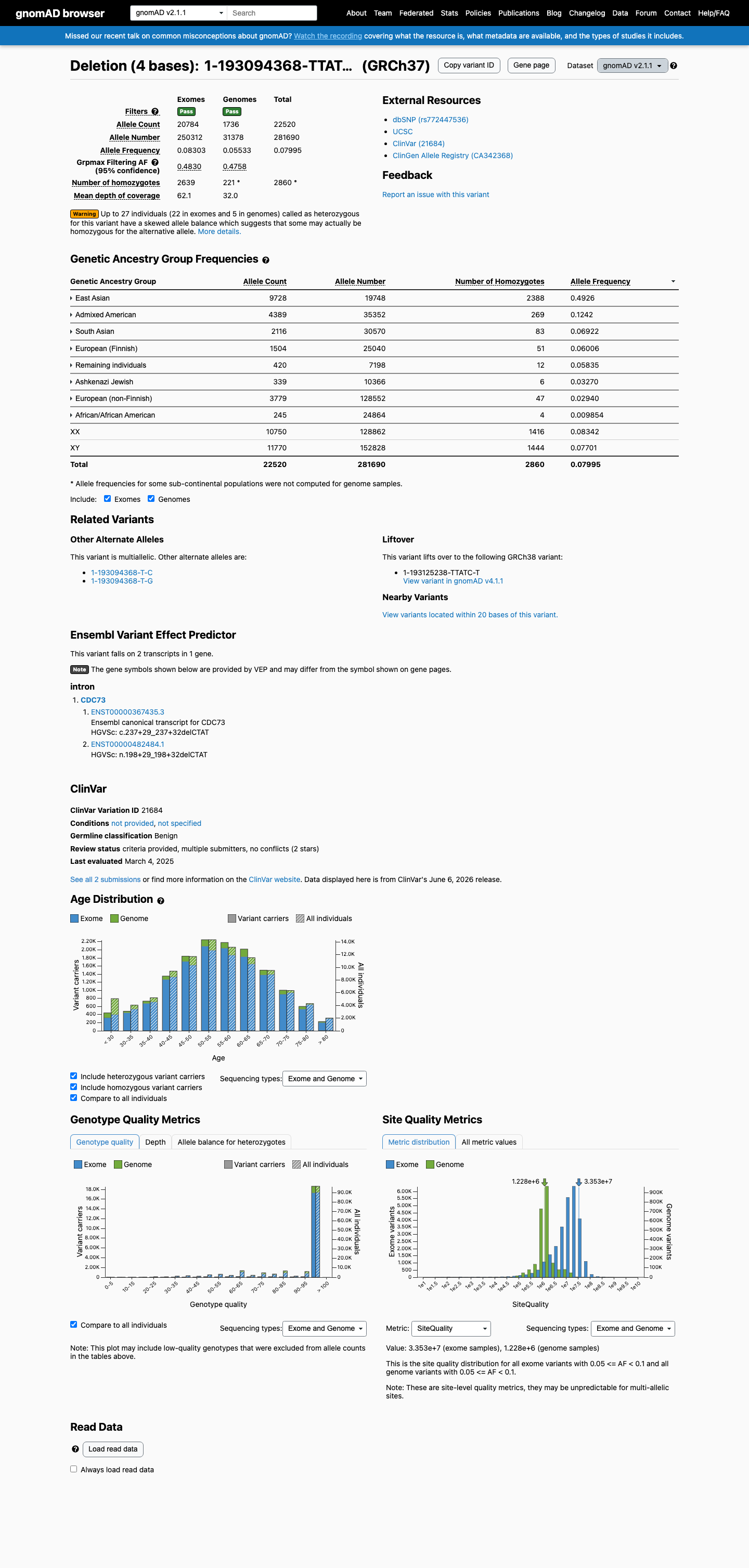
This variant has an allele frequency of 7.99% in gnomAD v2.1 (22,520/281,690 alleles, 2,860 homozygotes) and 4.74% in gnomAD v4.1 (63,137/1,331,598 alleles, 5,691 homozygotes), with a highest subpopulation frequency of 49.26% in East Asians, far exceeding the BA1 stand-alone benign threshold of >1%.1 The variant is observed in 2,860 homozygotes in gnomAD v2.1 and 5,691 homozygotes in gnomAD v4.1. CDC73 is associated with autosomal dominant hyperparathyroidism-jaw tumor syndrome with high penetrance; the presence of thousands of homozygous individuals in population databases is incompatible with pathogenicity (BS2).2 SpliceAI predicts no splice impact (max delta score = 0.00), consistent with a benign intronic variant (BP4).3 ClinVar reports this variant as Benign by two clinical laboratories using criteria-based assessment (BP6).4
CDC73
Final classification
Benign
CDC73 c.237+29_237+32del · p.?
CDC73
This variant has an allele frequency of 7.99% in gnomAD v2.1 (22,520/281,690 alleles, 2,860 homozygotes) and 4.74% in gnomAD v4.1 (63,137/1,331,598 alleles, 5,691 homozygotes), with a highest subpopulation frequency of 49.26% in East Asians, far exceeding the BA1 stand-alone benign threshold of >1%.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BA1 stand-alone benign, BS1 strong benign, BS2 strong benign, BP4 supporting benign, BP6 supporting benign; combination = 1 stand-alone benign + 2 strong benign + 2 supporting benign, which maps to Benign.
Classification rationale
BA1BS1BS2BP4BP6
Benign
CDC73 c.237+29_237+32del
BA1 + BS1 + BS2 + BP4 + BP6
→
Benign
Gene diagram
· NM_024529.5 · variants mapped to exon structure
CDC73
NM_024529.5
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 2 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
BA1
stand-alone
Benign
This variant has an allele frequency of 7.99% in gnomAD v2.1 (22,520/281,690 alleles, 2,860 homozygotes) and 4.74% in gnomAD v4.1 (63,137/1,331,598 alleles, 5,691 homozygotes), with a highest subpopulation frequency of 49.26% in East Asians. These frequencies far exceed the BA1 threshold of >1%. The variant is a common population polymorphism.
gnomAD v2.1: overall AF = 7.99% (22520/281690 alleles
✓
BS1
strong
Benign
This variant has an allele frequency of 7.99% in gnomAD v2.1, exceeding the BS1 threshold of >0.3%. This evidence is superseded by BA1 (stand-alone benign).
gnomAD v2.1 overall AF = 7.99%exceeding BS1 threshold of >0.3%.Superseded by BA1.
✓
BS2
strong
Benign
This variant is observed in 2,860 homozygotes in gnomAD v2.1 and 5,691 homozygotes in gnomAD v4.1. CDC73 is associated with autosomal dominant hyperparathyroidism-jaw tumor syndrome (HPT-JT) and parathyroid carcinoma, disorders with high penetrance. The observation of thousands of apparently healthy homozygous adults is incompatible with a pathogenic role.
gnomAD v2.1: 2860 homozygotesgnomAD v4.1: 5
✓
BP4
supporting
Benign
Multiple lines of computational evidence suggest no impact on splicing. SpliceAI predicts no splice impact (max delta score = 0.00) for this intronic deletion. No in silico predictors suggest a deleterious effect on gene or gene product.
SpliceAI max delta = 0.00no predicted donor/acceptor gain or loss.No in silico evidence supporting a deleterious effect.
✓
BP6
supporting
Benign
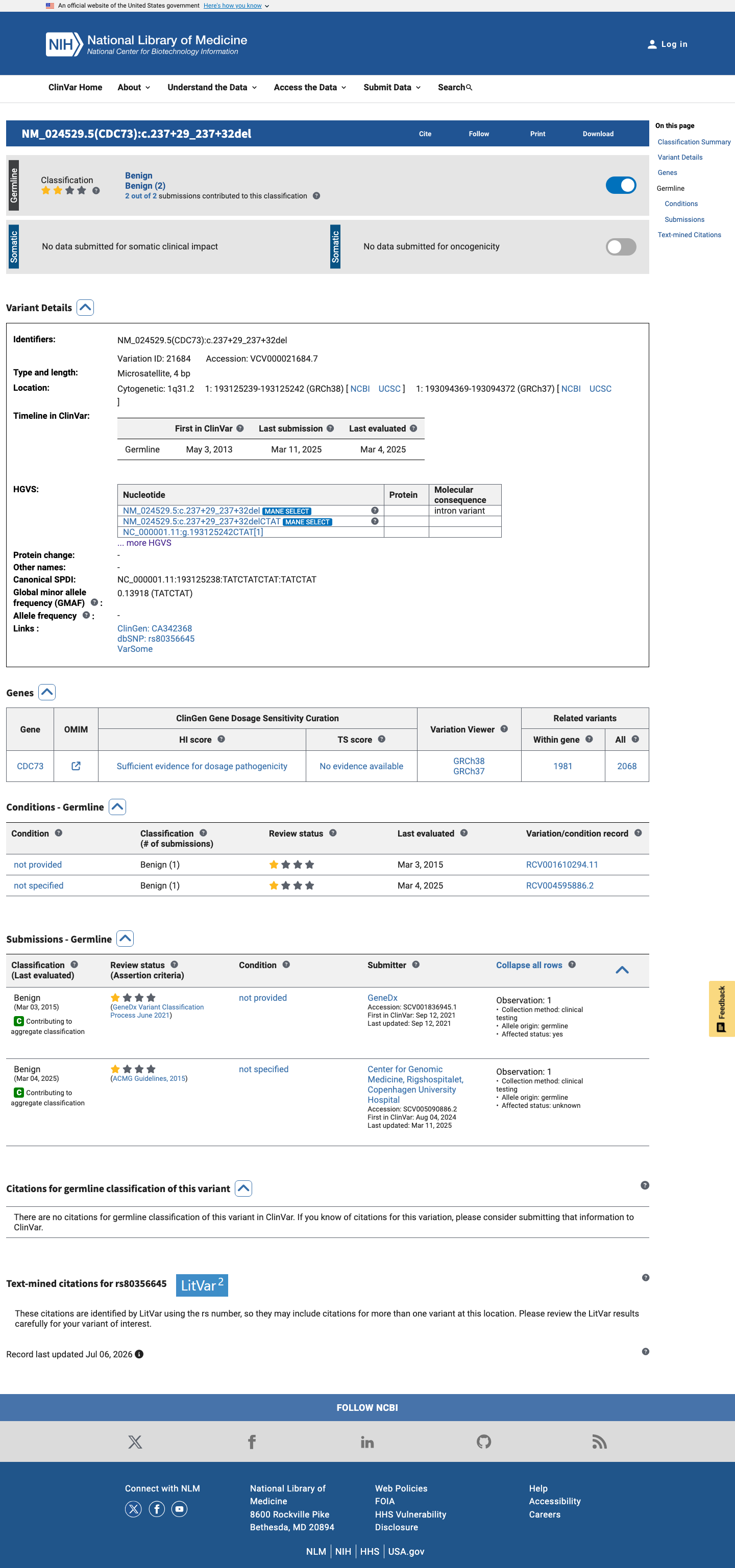
ClinVar reports this variant as Benign by two clinical laboratories (GeneDx and Center for Genomic Medicine, Rigshospitalet). Both submissions used criteria-based assessment in a clinical testing context.
ClinVar classification: Benign (ClinVar ID 21684).Two clinical laboratories: GeneDx (SCV001836945) and Center for Genomic MedicineRigshospitalet (SCV005090886).
Assessed · not applied
Pathogenic
PM2
This variant is present at high frequency in population databases: gnomAD v2.1 AF = 7.99% (22,520/281,690 alleles, 2,860 homozygotes) and gnomAD v4.1 AF = 4.74% (63,137/1,331,598 alleles, 5,691 homozygotes).
PP3
Multiple lines of computational evidence do not support a deleterious effect.
Benign
None assessed.
N/A · 20
PVS1 · PS1 · PS2 · PS3 · PS4 · PM1 · PM4 · PM5 · PM6 · PP1 · PP2 · PP4 · PP5 · BS3 · BS4 · BP1 · BP2 · BP3 · BP5 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.0474145; MAF= 4.74145%, 63137/1331598 alleles, homozygotes = 5691) and has highest observed frequency in the East Asian population (AF= 0.443013; MAF= 44.30127%, 19116/43150 alleles, homozygotes = 4351); grpmax FAF= 0.437755.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.079946; MAF= 7.99460%, 22520/281690 alleles, homozygotes = 2860) and has highest observed frequency in the East Asian population (AF= 0.492607; MAF= 49.26068%, 9728/19748 alleles, homozygotes = 2388); grpmax FAF= 0.483021.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.07179320082545888, 1322/18414 alleles, homozygotes = 174).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
4.7%
· 63137 / 1,331,598
5691 hom · FAF 44%
5691 hom · FAF 44%
East Asian 19116 / 43,150 |
44% 4351 hom |
Admixed American 6484 / 59,628 |
11% 389 hom |
South Asian 5902 / 85,206 |
6.9% 265 hom |
European (Finnish) 3568 / 63,846 |
5.6% 110 hom |
Remaining individuals 2952 / 53,218 |
5.5% 202 hom |
Middle Eastern 233 / 5,504 |
4.2% 7 hom |
Ashkenazi Jewish 888 / 27,898 |
3.2% 9 hom |
European (non-Finnish) 23360 / 922,740 |
2.5% 353 hom |
African/African American 634 / 69,496 |
0.91% 5 hom |
+ 1 not observed (Amish)
gnomAD v2.1
8%
· 22520 / 281,690
2860 hom · FAF 48%
2860 hom · FAF 48%
East Asian 9728 / 19,748 |
49% 2388 hom |
Admixed American 4389 / 35,352 |
12% 269 hom |
South Asian 2116 / 30,570 |
6.9% 83 hom |
European (Finnish) 1504 / 25,040 |
6% 51 hom |
Remaining individuals 420 / 7,198 |
5.8% 12 hom |
Ashkenazi Jewish 339 / 10,366 |
3.3% 6 hom |
European (non-Finnish) 3779 / 128,552 |
2.9% 47 hom |
African/African American 245 / 24,864 |
0.99% 4 hom |
gnomAD Canada 🇨🇦
7.2%
· 1322 / 18,414
174 hom · FAF 46%
174 hom · FAF 46%
indel · split
East Asian 652 / 1,336 |
49% 163 hom |
European (Finnish) 1 / 8 |
12% |
Latino/Admixed American 69 / 836 |
8.3% 3 hom |
Remaining individuals 93 / 1,136 |
8.2% 5 hom |
South Asian 85 / 1,360 |
6.2% 1 hom |
Middle Eastern 6 / 144 |
4.2% |
Ashkenazi Jewish 30 / 832 |
3.6% 1 hom |
European (non-Finnish) 379 / 11,742 |
3.2% 1 hom |
African/African American 7 / 1,020 |
0.69% |
ClinVar
This variant has been reported in ClinVar as Benign (2 clinical laboratories). (ClinVarID = 21684)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
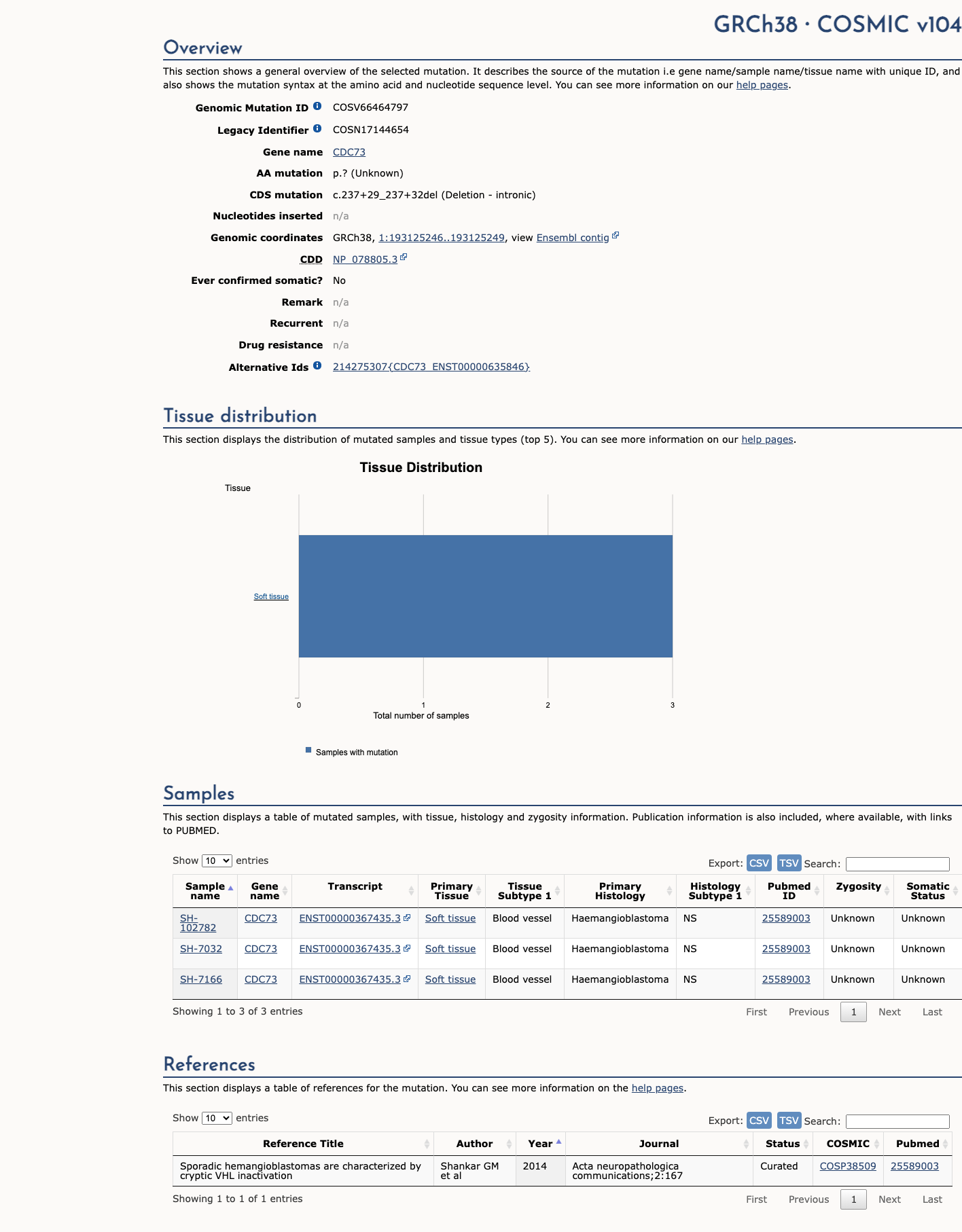
COSMIC
Somatic evidence
COSMIC
This variant has previously been reported in somatic cancers (COSMIC; COSV66464797, n = 3 times).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 1 PMID not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR