NM_032043.3:c.1794+19T>C is a deep intronic substitution in BRIP1 intron 12 with no predicted effect on splicing (SpliceAI max delta = 0.10).1 The variant is extremely rare in population databases (gnomAD v2.1 AF = 0.00389%, v4.1 AF = 0.00285%), meeting PM2 at supporting level.2 Multiple lines of computational evidence predict no deleterious splicing effect, meeting BP4 at supporting benign level.3 ClinVar reports Likely benign classification by 3 clinical laboratories (GeneDx, Color Health, Labcorp/Invitae), though the 1-star review status does not independently meet BP6 at supporting level under current PP5/BP6 guidance.4 No functional studies, case-control data, cosegregation data, de novo reports, or variant-specific literature exist for this variant. With one supporting pathogenic criterion (PM2) and one supporting benign criterion (BP4), the evidence is balanced and insufficient to classify as either Likely Pathogenic or Likely Benign. The variant is classified as a Variant of Uncertain Significance (VUS) under generic ACMG/AMP 2015 combination rules.5
BRIP1
Final classification
VUS
BRIP1 c.1794+19T>C · p.?
BRIP1
NM_032043.3:c.1794+19T>C is a deep intronic substitution in BRIP1 intron 12 with no predicted effect on splicing (SpliceAI max delta = 0.10).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting benign; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP4
VUS
BRIP1 c.1794+19T>C
PM2 + BP4
→
VUS
Gene diagram
· NM_032043.3 · variants mapped to exon structure
BRIP1
NM_032043.3
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 16 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_032043.3:c.1794+19T>C is present at extremely low frequency in gnomAD (v2.1 AF = 0.00389%, 11/282,822 alleles; v4.1 AF = 0.00285%, 46/1,612,392 alleles), well below the 0.1% PM2 threshold. The variant is absent from most subpopulations, has 0 homozygotes, and has a grpmax FAF of 1.122e-05. Observed primarily in the Ashkenazi Jewish population (ASJ AF = 0.058%) but remains rare even in this subpopulation.
gnomAD v2.1: AF = 3.89e-05 (0.00389%)11/282822 alleles
✓
BP4
supporting
Benign
Multiple lines of computational evidence support a benign interpretation. SpliceAI predicts no significant splicing impact for NM_032043.3:c.1794+19T>C (max delta score = 0.10). The variant is located at intron position +19, well outside the canonical splice donor/acceptor consensus. No in silico tool predicts a deleterious effect on splicing.
SpliceAI max delta = 0.10 across all categories (acceptor gainacceptor lossdonor gain
Assessed · not applied
Pathogenic
PS2
No de novo evidence is available for NM_032043.3:c.1794+19T>C.
PS3
No functional studies have been performed on NM_032043.3:c.1794+19T>C.
PS4
No case-control or case series data are available for NM_032043.3:c.1794+19T>C.
PM6
No de novo evidence is available for NM_032043.3:c.1794+19T>C.
PP1
No cosegregation data are available for NM_032043.3:c.1794+19T>C.
PP3
Multiple lines of computational evidence do not support a deleterious effect.
PP4
No patient phenotype or clinical data are available to evaluate whether the proband's presentation is specific for BRIP1-related disease.
PP5
ClinVar classification for NM_032043.3:c.1794+19T>C is Likely benign (Variation ID 377594), not pathogenic.
Benign
BA1
NM_032043.3:c.1794+19T>C has a gnomAD v2.1 total allele frequency of 0.00389% (3.89e-05), which is far below the 1% BA1 threshold for a benign standing variant.
BS1
NM_032043.3:c.1794+19T>C has a gnomAD v2.1 total allele frequency of 0.00389%, which is below the 0.3% BS1 threshold under generic ACMG rules.
BS2
No evidence that NM_032043.3:c.1794+19T>C has been observed in a healthy adult individual as homozygous or in trans with a known pathogenic BRIP1 variant.
BS3
No functional studies demonstrating no deleterious effect have been performed for NM_032043.3:c.1794+19T>C.
BS4
No cosegregation data are available for NM_032043.3:c.1794+19T>C.
BP2
No data are available on whether NM_032043.3:c.1794+19T>C has been observed in trans with a known pathogenic BRIP1 variant.
BP5
No information is available regarding an alternate molecular basis for disease in the proband.
BP6
Three clinical laboratories (GeneDx, Color Health, Labcorp/Invitae) independently classify NM_032043.3:c.1794+19T>C as Likely benign in ClinVar (Variation ID 377594).
N/A · 10
PVS1 · PS1 · PM1 · PM3 · PM4 · PM5 · PP2 · BP1 · BP3 · BP7
Research & evidence
Population frequency
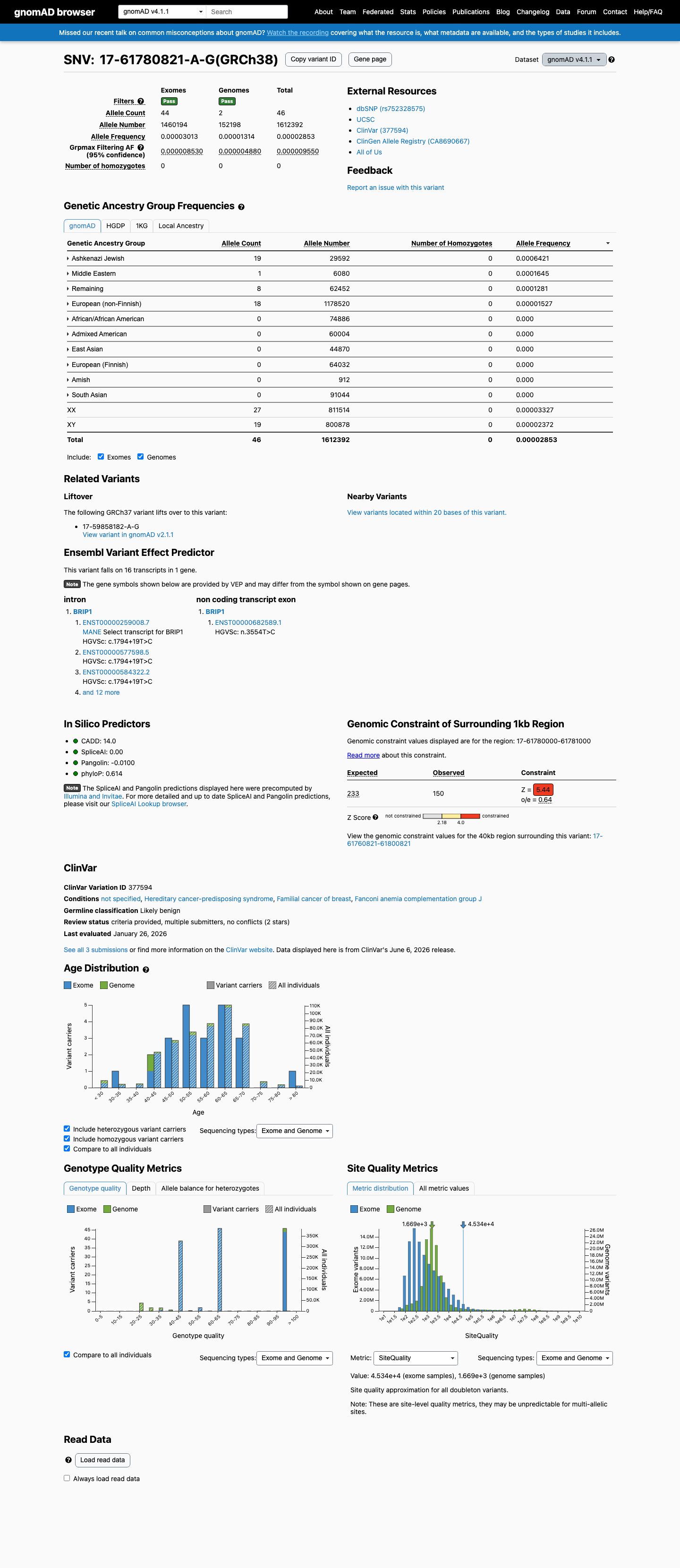
gnomAD v4.1
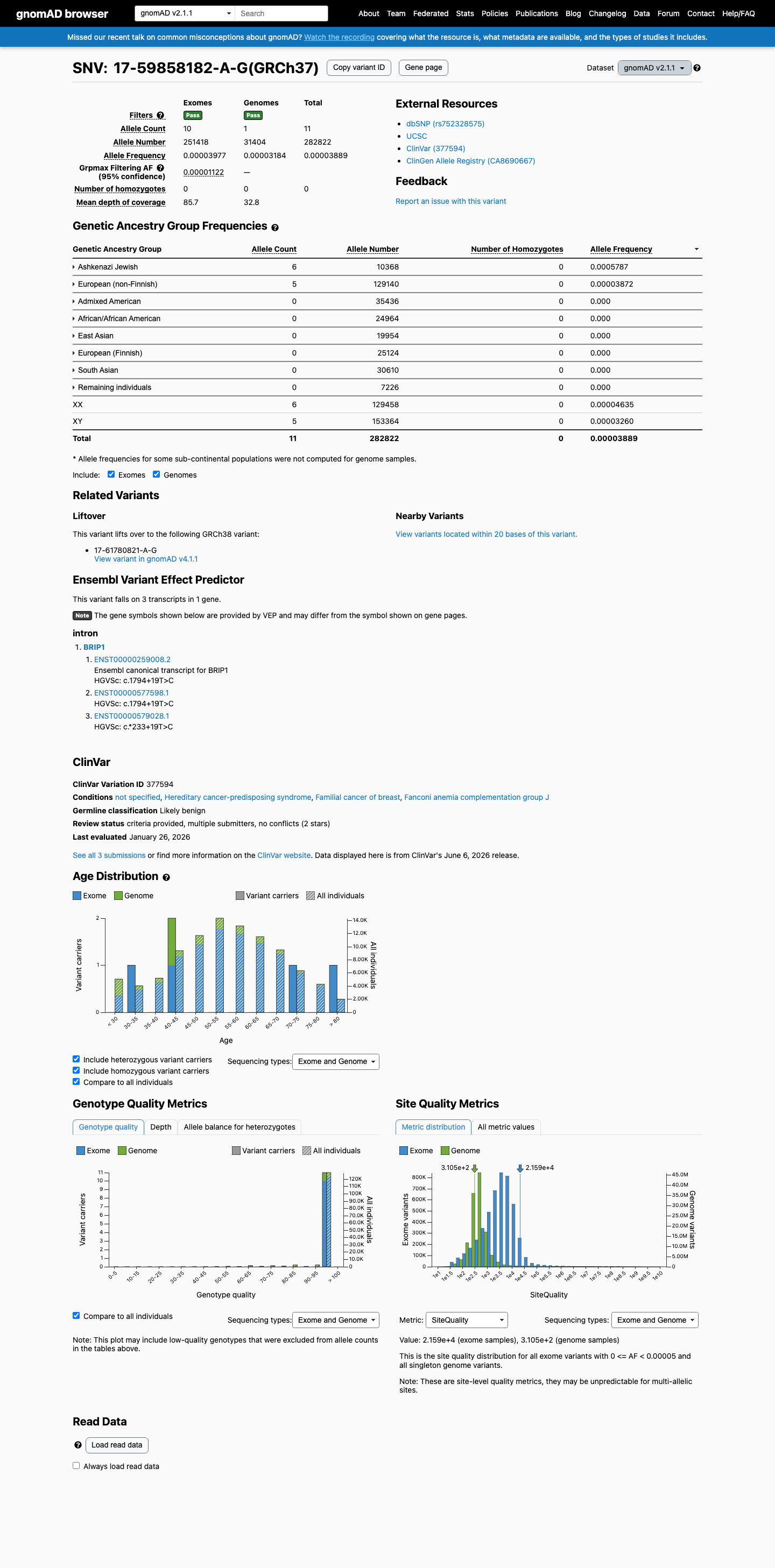
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 2.8529e-05; MAF= 0.00285%, 46/1612392 alleles, homozygotes = 0) and has highest observed frequency in the Ashkenazi Jewish population (AF= 0.000642065; MAF= 0.06421%, 19/29592 alleles, homozygotes = 0); grpmax FAF= 9.55e-06.
v2.1
This variant is present in gnomAD v2.1 (AF= 3.88937e-05; MAF= 0.00389%, 11/282822 alleles, homozygotes = 0) and has highest observed frequency in the Ashkenazi Jewish population (AF= 0.000578704; MAF= 0.05787%, 6/10368 alleles, homozygotes = 0); grpmax FAF= 1.122e-05.
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0029%
· 46 / 1,612,392
0 hom · FAF 0.00096%
0 hom · FAF 0.00096%
Ashkenazi Jewish 19 / 29,592 |
0.064% |
Middle Eastern 1 / 6,080 |
0.016% |
Remaining individuals 8 / 62,452 |
0.013% |
European (non-Finnish) 18 / 1,178,520 |
0.0015% |
+ 6 not observed (Admixed American, European (Finnish), Amish, East Asian, South Asian, African/African American)
gnomAD v2.1
0.0039%
· 11 / 282,822
0 hom · FAF 0.0011%
0 hom · FAF 0.0011%
Ashkenazi Jewish 6 / 10,368 |
0.058% |
European (non-Finnish) 5 / 129,140 |
0.0039% |
+ 6 not observed (African/African American, Admixed American, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
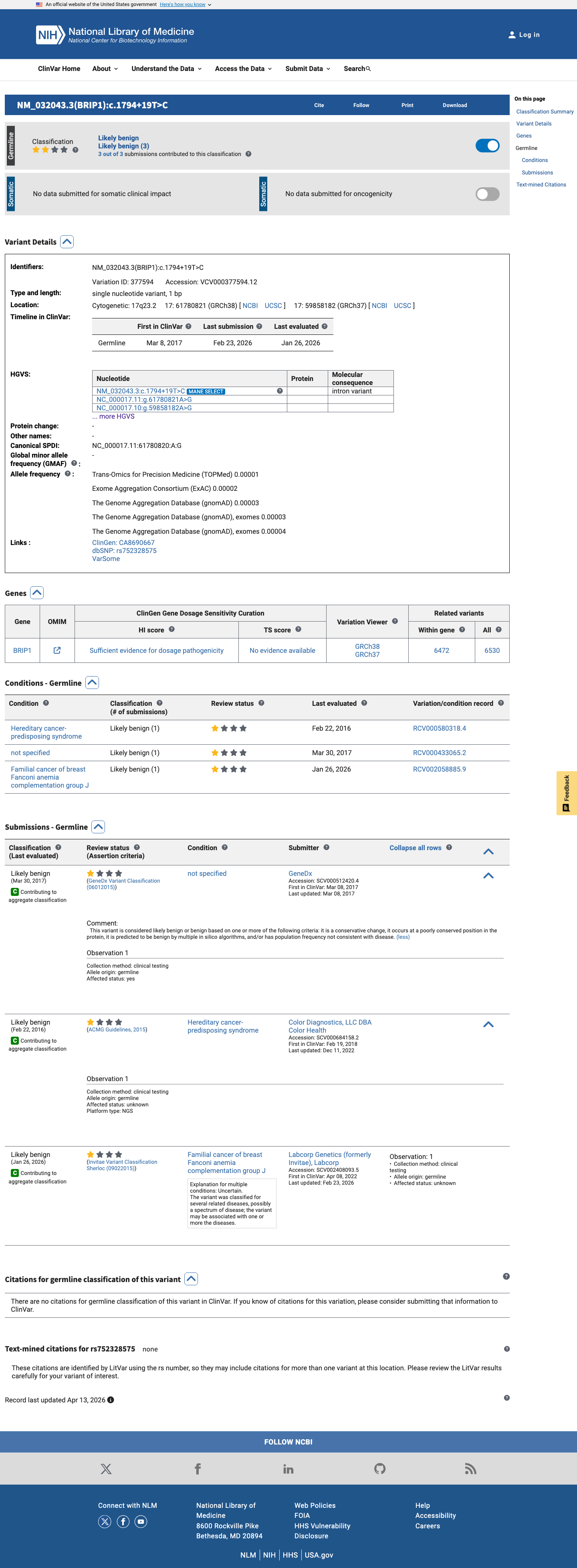
ClinVar
This variant has been reported in ClinVar as Likely benign (3 clinical laboratories). (ClinVarID = 377594)
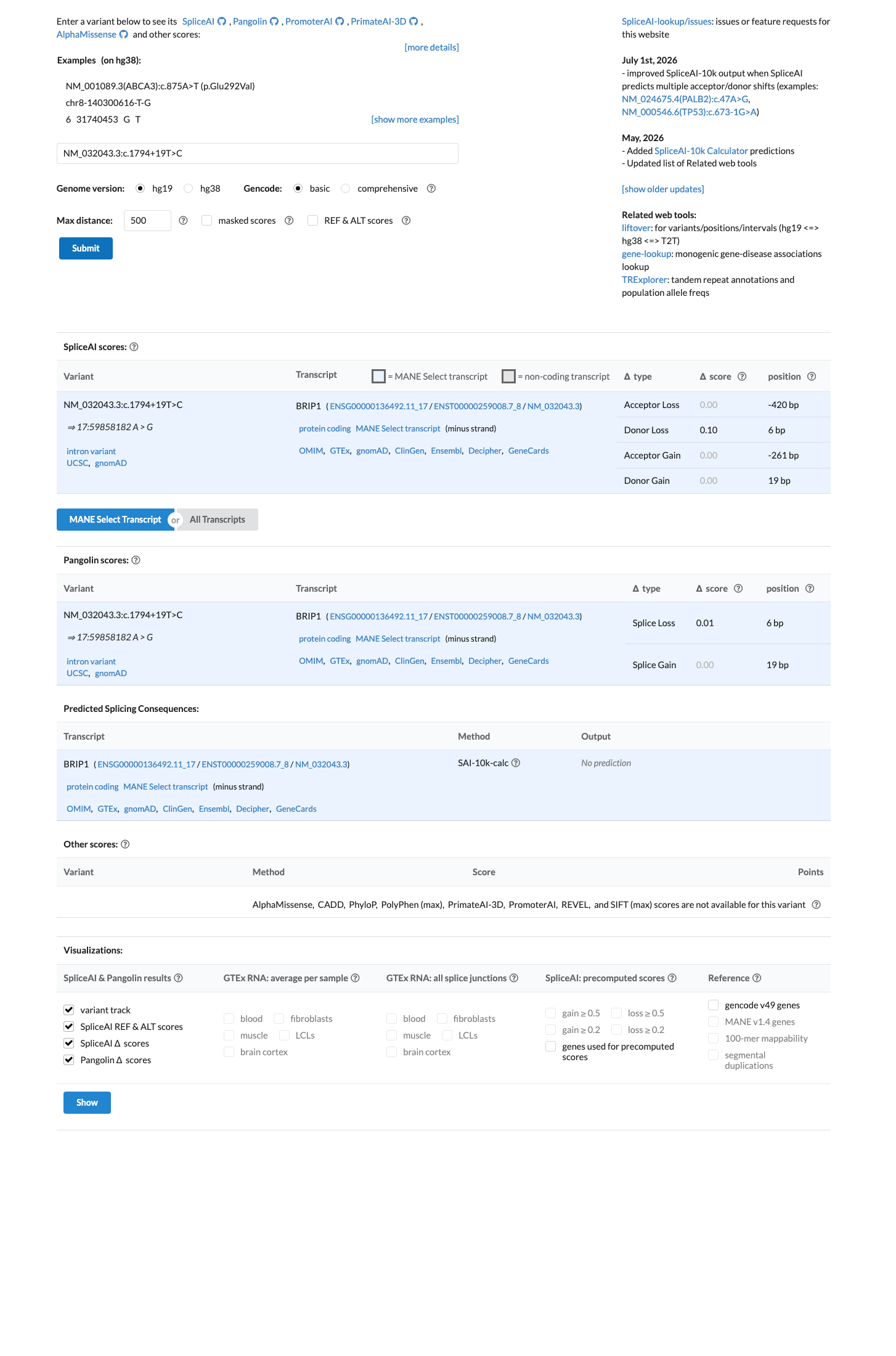
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.10).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 7 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
24366376 ↗
Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women: U.S. Preventive Services Task Force recommendation statement.
CLINVAR
24366402 ↗
Summaries for patients. Assessing the genetic risk for BRCA-related breast or ovarian cancer in women: recommendations from the U.S. Preventive Services Task Force.
CLINVAR
31429903 ↗
Risk Assessment, Genetic Counseling, and Genetic Testing for BRCA-Related Cancer: US Preventive Services Task Force Recommendation Statement.
CLINVAR
35802134 ↗
ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG).
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR