Classification rationale
BA1BS1BP4
Benign
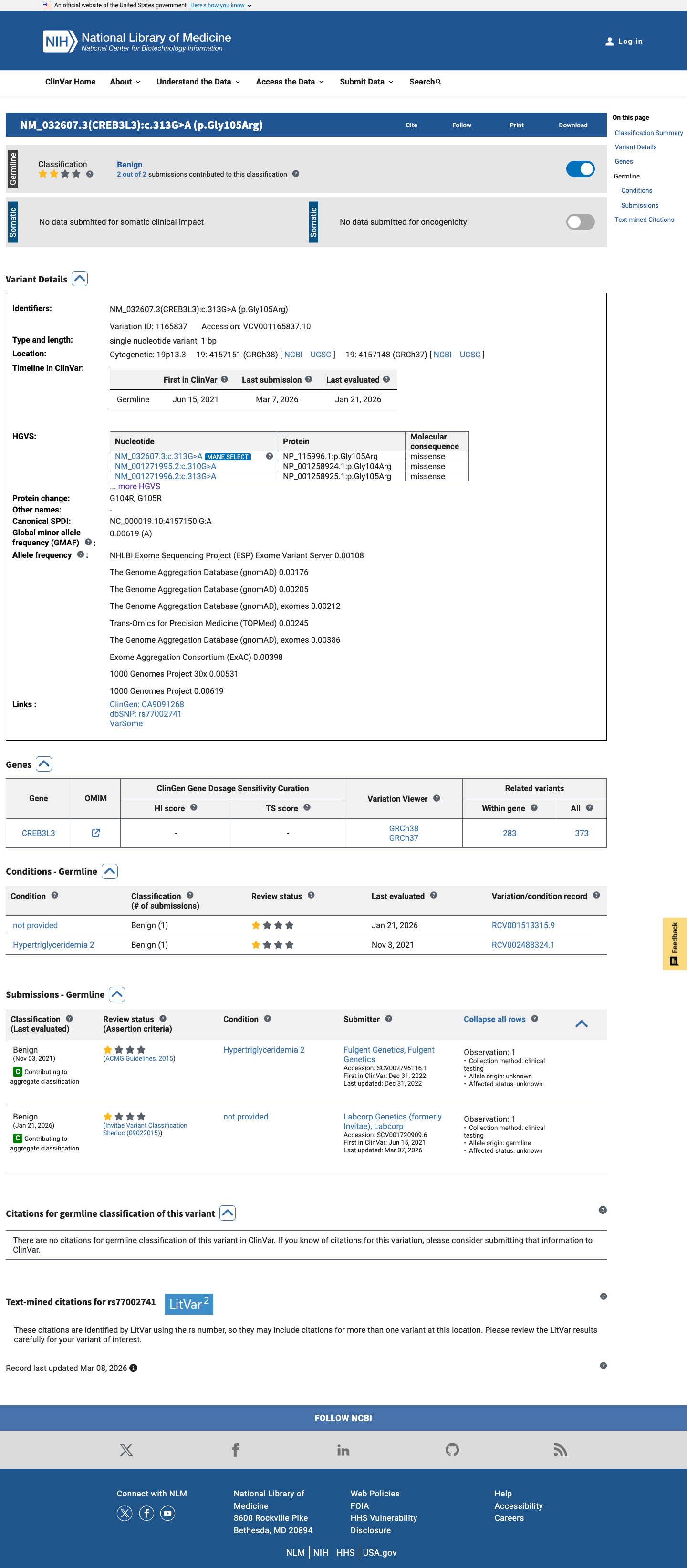
CREB3L3 c.313G>A
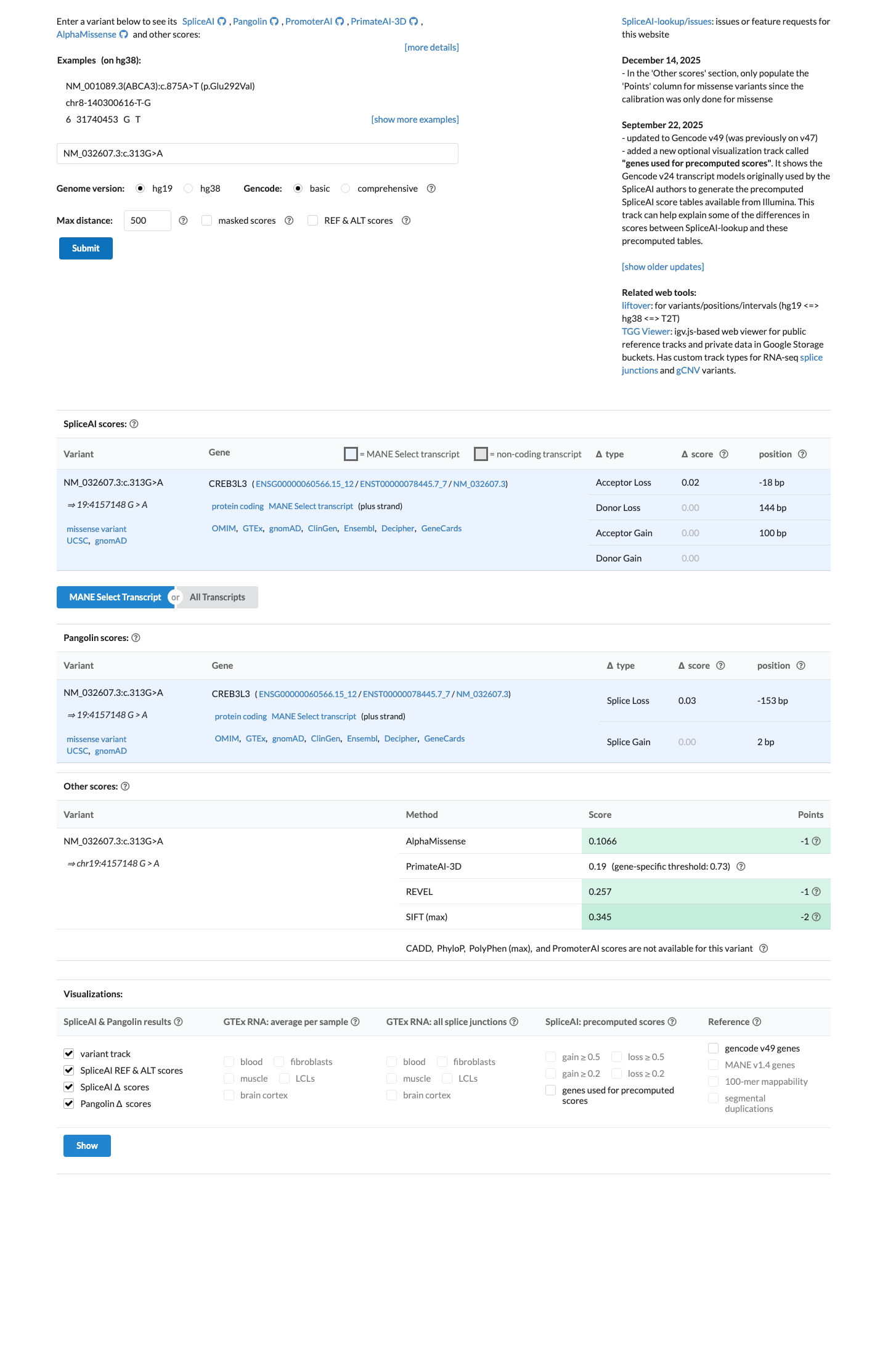
The CREB3L3 c.313G>A (p.Gly105Arg, p.G105R) variant has been reported in ClinVar as Benign with criteria provided by a single submitter representing 2 clinical laboratory submissions.1 This variant is common in population databases, with gnomAD v2.1 showing a total allele frequency of 0.37177% and an East Asian allele frequency of 3.44274%, and gnomAD v4.1 showing a total allele frequency of 0.21551% and an East Asian allele frequency of 3.43413%, which is above the default benign population thresholds.2 Computational evidence does not support a damaging effect, with SpliceAI predicting no significant splice impact (maximum delta score 0.02), REVEL 0.257, and BayesDel -0.423809.3
BA1 + BS1 + BP4
→
Benign