NM_080605.4:c.901_904dup is a frameshift variant in B3GALT6 predicted to alter the C-terminus (p.Arg302GlnfsTer142). B3GALT6 loss of function is a well-established disease mechanism for autosomal recessive spondylodysplastic Ehlers-Danlos syndrome. PVS1 is applied at strong strength, downgraded from very_strong due to the 3' location (codon 302/330) and confirmed NMD escape in this single-exon gene (PMC6185798).1 The variant is extremely rare in population databases, meeting PM2_Supporting: gnomAD v2.1 allele frequency 7.17e-06 (1/139,512 alleles) and gnomAD v4.1 allele frequency 9.06e-06 (14/1,544,998 alleles), both well below the 0.1% threshold. No homozygotes are observed.2 The variant is present in ClinVar (Variation ID 1323966) with four clinical testing submissions: two classify it as Likely Pathogenic and two as Uncertain Significance. No expert panel review is available, and the cited PubMed references (PMID:25741868, PMID:28492532) are methodology guidelines that do not specifically mention this variant.3 SpliceAI predicts no significant splicing impact (max delta score 0.03). In silico missense predictors (REVEL, BayesDel) are not applicable to this frameshift variant. PP3 is not met; BP4 is not met given insufficient benign computational evidence.4 Applying generic ACMG/AMP 2015 final classification rules (PMID:25741868): with one strong pathogenic criterion (PVS1_Strong) and one supporting pathogenic criterion (PM2_Supporting), the variant does not reach the threshold for Likely Pathogenic (requires 1 Strong + 1–2 Moderate OR 1 Strong + 2 Supporting). The variant is classified as a Variant of Uncertain Significance (VUS). Additional evidence, particularly verified publication reports of the variant in affected individuals (PS4/PM3) and segregation data (PP1), may upgrade the classification upon human curator review.5
B3GALT6
Final classification
VUS
B3GALT6 c.901_904dup · p.Arg302GlnfsTer142
B3GALT6
NM_080605.4:c.901_904dup is a frameshift variant in B3GALT6 predicted to alter the C-terminus (p.Arg302GlnfsTer142). B3GALT6 loss of function is a well-established disease mechanism for autosomal recessive spondylodysplastic Ehlers-Danlos syndrome. PVS1 is applied at strong strength, downgraded from very_strong due to the 3' location (codon 302/330) and confirmed NMD escape in this single-exon gene (PMC6185798).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 strong, PM2 supporting; combination = 1 strong + 1 supporting, which maps to VUS.
Classification rationale
PVS1PM2
VUS
B3GALT6 c.901_904dup
PVS1 + PM2
→
VUS
1
pvs1_gene_contextpvs1_variant_assessmentpvs1_generic_framework ↗
5
generic_acmg_combination_rulesPMID:25741868 ↗
Gene diagram
· NM_080605.4 · variants mapped to exon structure
B3GALT6
NM_080605.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 17 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
strong
Pathogenic
This duplication (c.901_904dup) is a frameshift variant predicted to result in a premature termination codon (p.Arg302GlnfsTer142). B3GALT6 loss of function is a well-established disease mechanism for autosomal recessive spondylodysplastic Ehlers-Danlos syndrome (spEDS-B3GALT6). However, NM_080605.4 is a single-exon transcript and the frameshift occurs at codon 302 of 330, near the 3' end; NMD is not expected to be triggered. Per the ClinGen SVI PVS1 framework (PMC6185798), PVS1 is downgraded from very_strong to strong due to confirmed NMD escape in a 3' region.
Frameshift variant c.901_904dup leading to p.Arg302GlnfsTer142B3GALT6 LoF is an established disease mechanism for spEDS-B3GALT6 (supported by PMID:23664118PMID:25331875
✓
PM2
supporting
Pathogenic
This variant is present at extremely low frequency in population databases: gnomAD v2.1 allele frequency 7.17e-06 (1/139,512 alleles, 0.00072%) and gnomAD v4.1 allele frequency 9.06e-06 (14/1,544,998 alleles, 0.00091%). Both are well below the 0.1% PM2 threshold. No homozygotes are reported. Absent from gnomAD-Canada v1.0.
gnomAD v2.1: AF=7.17e-06 (1/1395120 homozygotes)
Assessed · not applied
Pathogenic
PS2
No de novo occurrence of NM_080605.4:c.901_904dup has been reported with confirmed maternity and paternity.
PS3
No functional studies evaluating the impact of NM_080605.4:c.901_904dup on B3GALT6 protein function were identified.
PS4
The variant prevalence in affected individuals versus the general population could not be reliably established.
PM1
No established mutational hotspot or critical functional domain data specific to B3GALT6 residue 302 or the C-terminal region are available to support PM1.
PM6
No report of a de novo occurrence of NM_080605.4:c.901_904dup exists in the literature or ClinVar submissions, even without confirmed maternity/paternity.
PP1
Cosegregation evidence could not be independently verified.
PP3
SpliceAI predicts no significant splice impact (max delta score = 0.03).
PP4
PP4 requires the patient's phenotype or family history to be highly specific for the disease with a single genetic etiology.
Benign
BA1
The highest population allele frequency is 0.00194% in the Admixed American population (gnomAD v4.1) and 0.00189% in European non-Finnish (gnomAD v2.1).
BS1
The highest population allele frequency is 0.00194% (gnomAD v4.1, AMR population).
BS2
BS2 requires observation of the variant in a healthy adult homozygous state or in trans with a pathogenic variant for a fully penetrant dominant disorder.
BS3
No functional studies demonstrating no deleterious effect of NM_080605.4:c.901_904dup were identified.
BS4
No evidence of non-segregation with disease has been reported.
BP2
BP2 requires observation in trans with a pathogenic variant for a fully penetrant dominant disorder, or in cis with a pathogenic variant.
BP4
BP4 requires multiple lines of computational evidence suggesting no impact on the gene or gene product.
BP5
BP5 requires an alternative molecular basis for disease to be identified in the case.
BP6
BP6 requires a reputable source to classify the variant as benign without independent verification.
N/A · 8
PS1 · PM4 · PM5 · PP2 · PP5 · BP1 · BP3 · BP7
Research & evidence
Population frequency
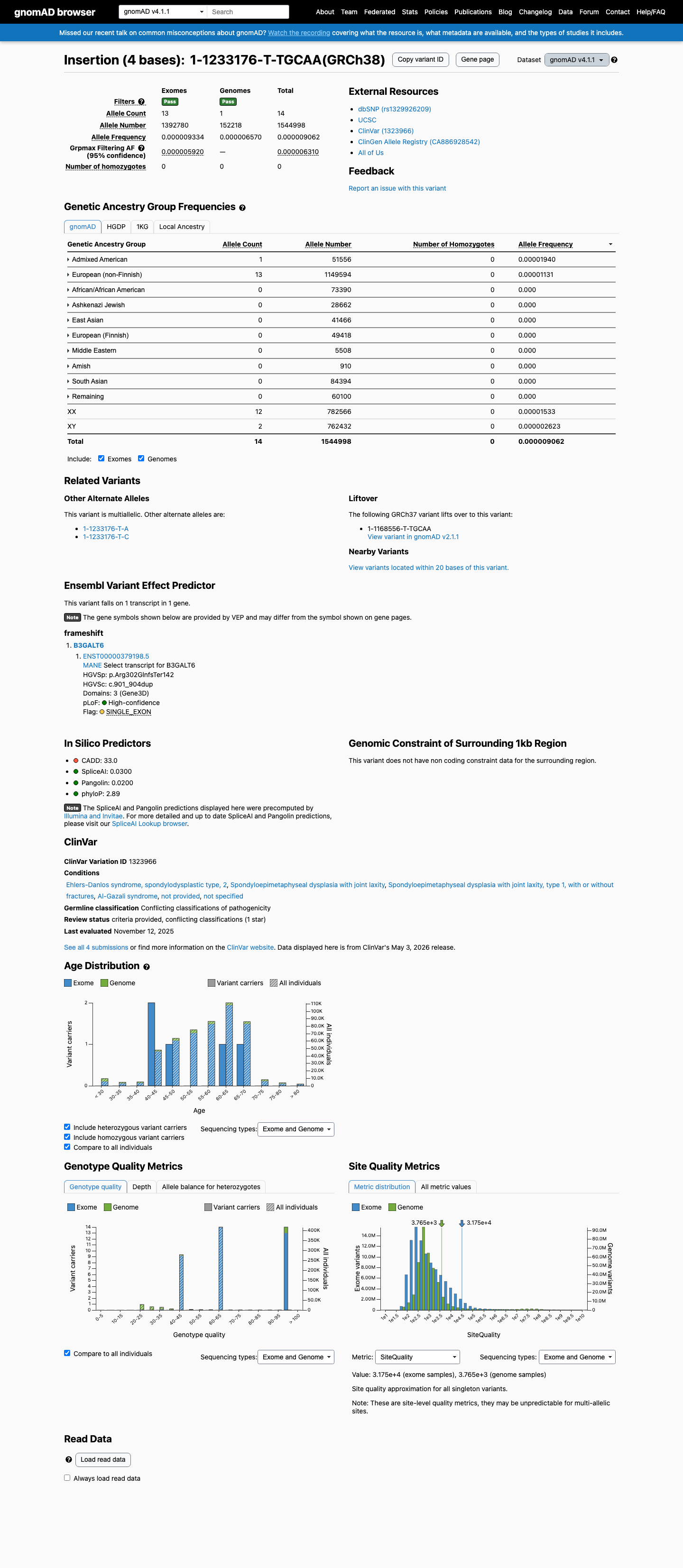
gnomAD v4.1
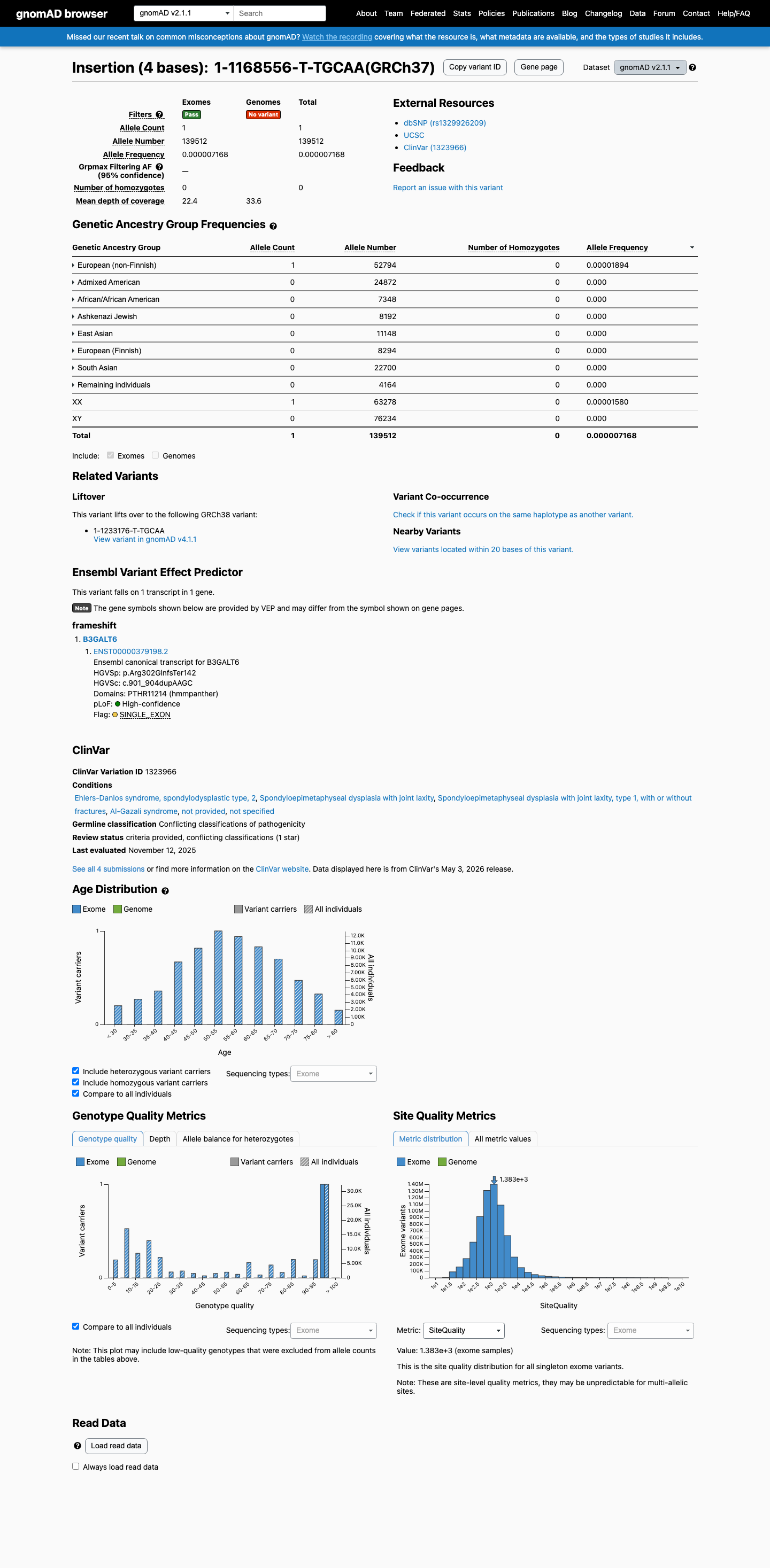
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 9.0615e-06; MAF= 0.00091%, 14/1544998 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 1.93964e-05; MAF= 0.00194%, 1/51556 alleles, homozygotes = 0); grpmax FAF= 6.31e-06.
v2.1
This variant is present in gnomAD v2.1 (AF= 7.16784e-06; MAF= 0.00072%, 1/139512 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 1.89415e-05; MAF= 0.00189%, 1/52794 alleles, homozygotes = 0).
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00091%
· 14 / 1,544,998
0 hom · FAF 0.00063%
0 hom · FAF 0.00063%
Admixed American 1 / 51,556 |
0.0019% |
European (non-Finnish) 13 / 1,149,594 |
0.0011% |
+ 8 not observed (Remaining individuals, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.00072%
· 1 / 139,512
0 hom
0 hom
European (non-Finnish) 1 / 52,794 |
0.0019% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
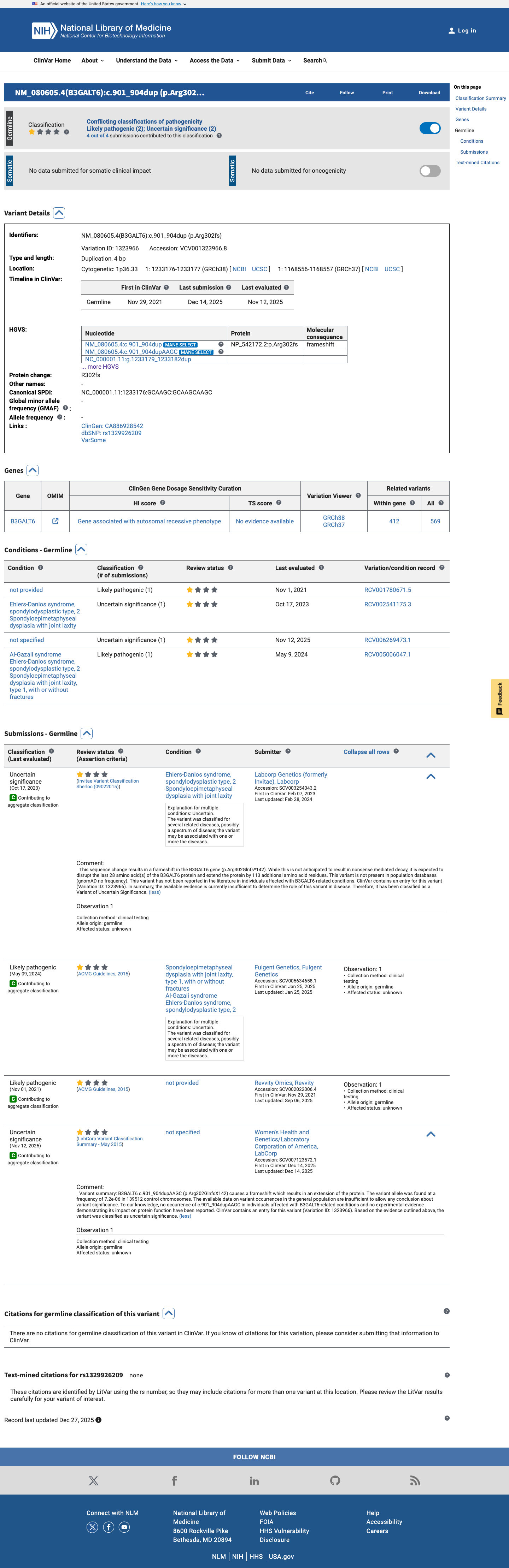
ClinVar
This variant is present in ClinVar (Variation ID: 1323966); submission details unavailable.
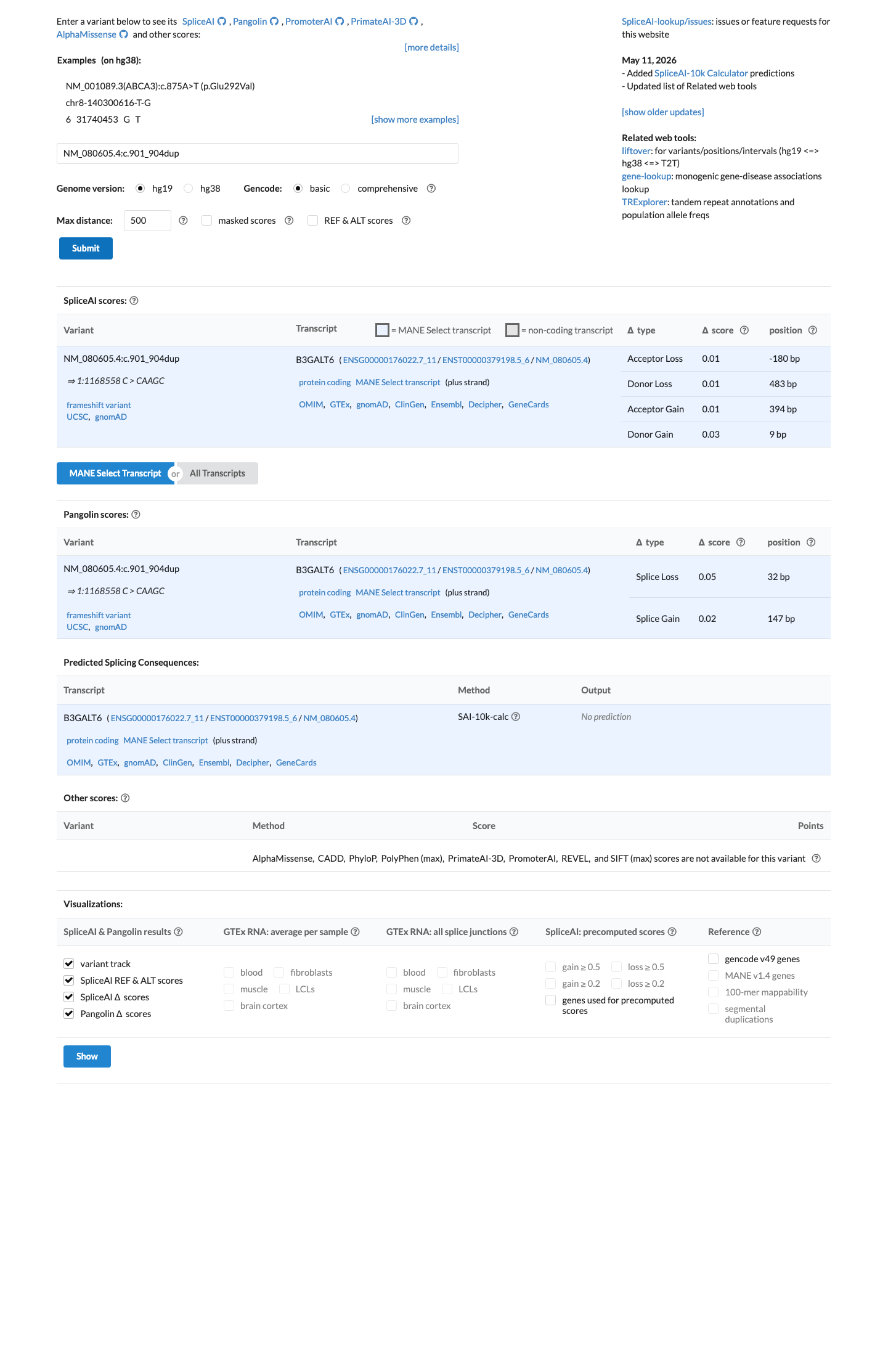
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.03).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
3papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 2 further PMIDs triaged but not cited — see Sources & References.
PMID 23664118
Found
Frameshift variant c.901_904dup leading to p.Arg302GlnfsTer142 B3GALT6 LoF is an established disease mechanism for spEDS-B3GALT6 (supported by PMID:23664118 PMID:25331875 PMID:38065100) Single-exon gene NMD escape predicted variant in 3' region (codon 302/330) ClinGen SVI PVS1 downgrade framework (PMC6185798) applied
Applied to
→PVS1 supports · met
PMID 25331875
Found
Frameshift variant c.901_904dup leading to p.Arg302GlnfsTer142 B3GALT6 LoF is an established disease mechanism for spEDS-B3GALT6 (supported by PMID:23664118 PMID:25331875 PMID:38065100) Single-exon gene NMD escape predicted variant in 3' region (codon 302/330) ClinGen SVI PVS1 downgrade framework (PMC6185798) applied
Applied to
→PVS1 supports · met
PMID 38065100
Found
Frameshift variant c.901_904dup leading to p.Arg302GlnfsTer142 B3GALT6 LoF is an established disease mechanism for spEDS-B3GALT6 (supported by PMID:23664118 PMID:25331875 PMID:38065100) Single-exon gene NMD escape predicted variant in 3' region (codon 302/330) ClinGen SVI PVS1 downgrade framework (PMC6185798) applied
Applied to
→PVS1 supports · met
Sources & reference links
Triaged references · 2 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR