The FLCN c.1333G>A (p.Ala445Thr) variant has been reported in ClinVar predominantly as likely benign or benign, with only a single uncertain significance submission.1 This variant is common in population databases, including gnomAD v2.1 with a total allele frequency of 0.25848% and a highest observed subpopulation frequency of 0.37088%, and gnomAD v4.1 with a total allele frequency of 0.29305% and a Middle Eastern population frequency of 1.35269%, which exceeds the usual benign stand-alone threshold of 1%.2 FLCN-related disease is primarily associated with loss-of-function variants, and the reviewed FLCN mutation database reported only 2 missense variants among 53 unique germline mutations, which supports a benign interpretation for this missense change.3 SpliceAI predicts no significant splice effect for this variant, with a maximum delta score of 0.00.4
FLCN
Final classification
Benign
FLCN c.1333G>A · p.Ala445Thr
FLCN
The FLCN c.1333G>A (p.Ala445Thr) variant has been reported in ClinVar predominantly as likely benign or benign, with only a single uncertain significance submission.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BA1 stand-alone benign, BP1 supporting; combination = 1 stand-alone benign + 1 supporting benign, which maps to Benign.
Classification rationale
BA1BP1
Benign
FLCN c.1333G>A
BA1 + BP1
→
Benign
Gene diagram
· NM_144997.7 · variants mapped to exon structure
FLCN
NM_144997.7
Fetching transcript structure from UCSC…
Applied criteria · 2 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency · supports benign
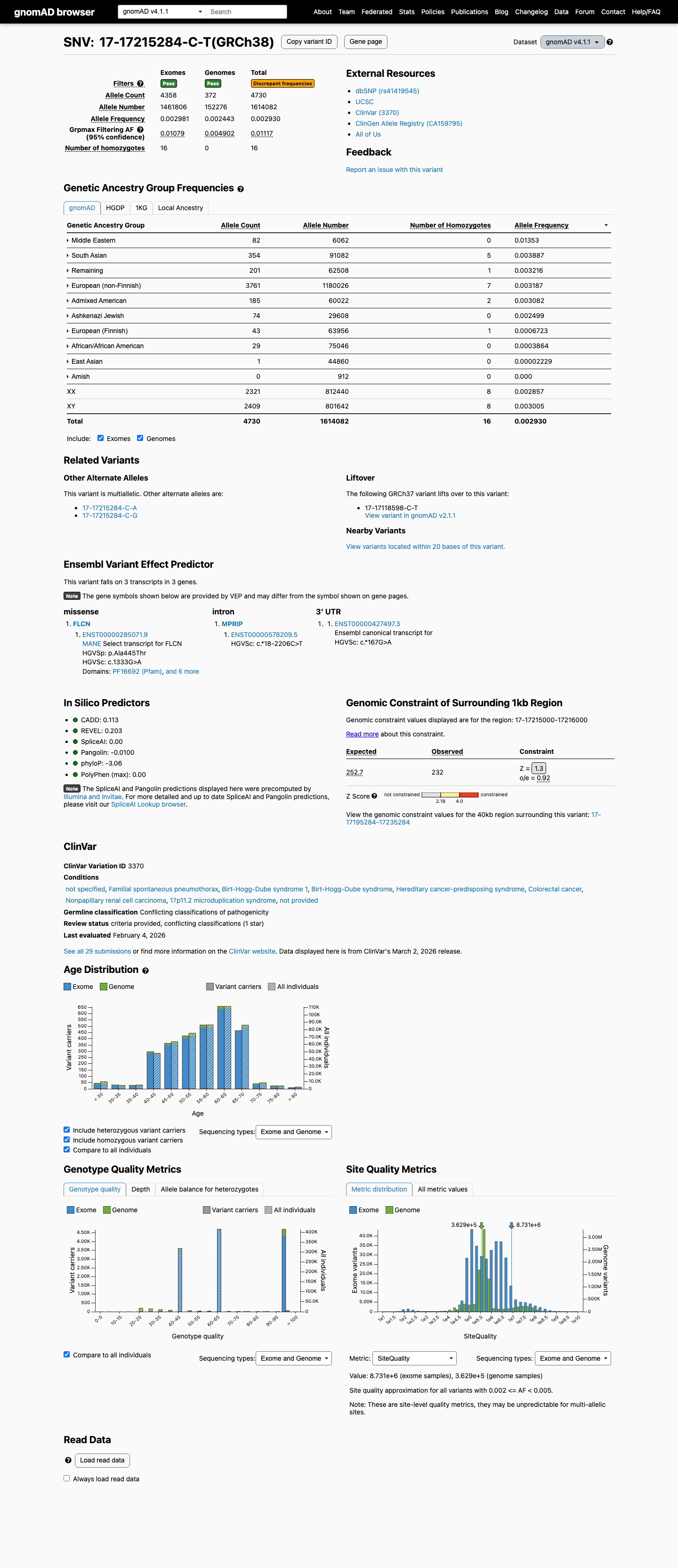
gnomAD v4.1

gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.00293046; MAF= 0.29305%, 4730/1614082 alleles, homozygotes = 16) and has highest observed frequency in the Middle Eastern population (AF= 0.0135269; MAF= 1.35269%, 82/6062 alleles, homozygotes = 0); grpmax FAF= 0.0111668.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.0025848; MAF= 0.25848%, 730/282420 alleles, homozygotes = 3) and has highest observed frequency in the European (non-Finnish) population (AF= 0.00370882; MAF= 0.37088%, 478/128882 alleles, homozygotes = 0); grpmax FAF= 0.00340865.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.29%
· 4730 / 1,614,082
16 hom · FAF 1.1%
16 hom · FAF 1.1%
Middle Eastern 82 / 6,062 |
1.4% |
South Asian 354 / 91,082 |
0.39% 5 hom |
Remaining individuals 201 / 62,508 |
0.32% 1 hom |
European (non-Finnish) 3761 / 1,180,026 |
0.32% 7 hom |
Admixed American 185 / 60,022 |
0.31% 2 hom |
Ashkenazi Jewish 74 / 29,608 |
0.25% |
European (Finnish) 43 / 63,956 |
0.067% 1 hom |
African/African American 29 / 75,046 |
0.039% |
East Asian 1 / 44,860 |
0.0022% |
+ 1 not observed (Amish)
gnomAD v2.1
0.26%
· 730 / 282,420
3 hom · FAF 0.34%
3 hom · FAF 0.34%
European (non-Finnish) 478 / 128,882 |
0.37% |
Remaining individuals 25 / 7,222 |
0.35% 1 hom |
South Asian 103 / 30,604 |
0.34% 1 hom |
Ashkenazi Jewish 28 / 10,350 |
0.27% |
Admixed American 72 / 35,430 |
0.2% 1 hom |
European (Finnish) 17 / 25,062 |
0.068% |
African/African American 7 / 24,924 |
0.028% |
+ 1 not observed (East Asian)
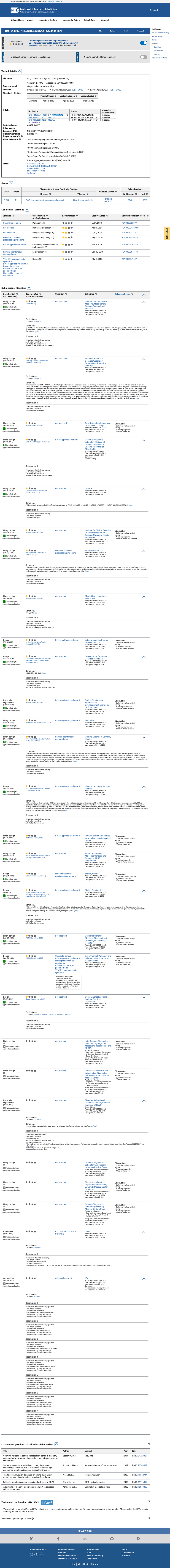
ClinVar
This variant has been reported in ClinVar as Likely benign (18 clinical laboratories) and as Benign (5 clinical laboratories) and as Uncertain significance (1 clinical laboratory) and as benign (1 clinical laboratory).
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.203. BayesDel score = -0.459533.
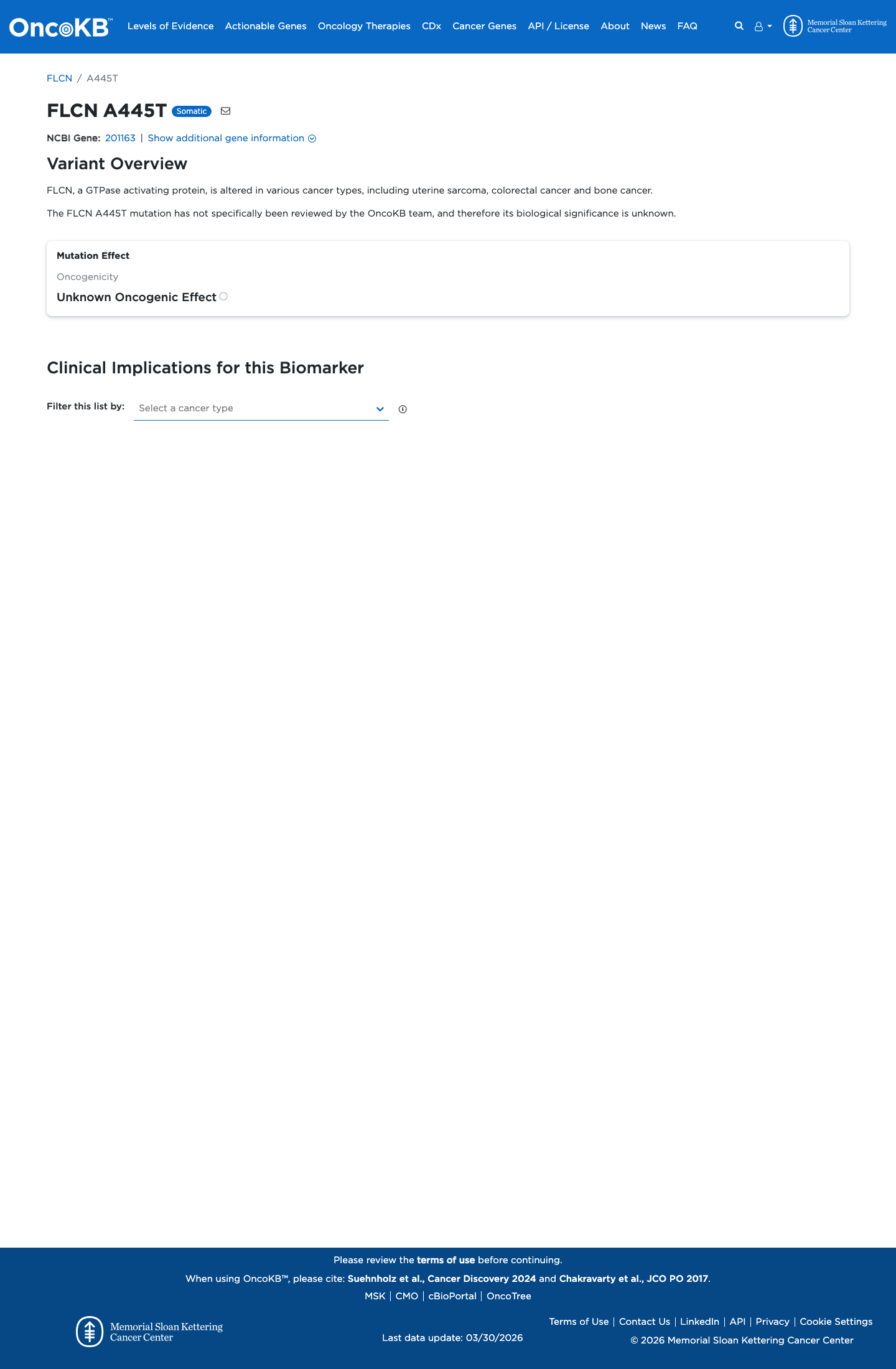
Functional
Unknown Oncogenic Effect
OncoKB has not reviewed this specific variant; no variant-level oncogenicity or biological effect is available. Gene-level context: FLCN, a GTPase activating protein, is altered in various cancer types, including uterine sarcoma, colorectal cancer and bone cancer.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV53262268, n = 2 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why.
PMID PMID:19562744
Found
Structured finding pending for this record — see source link.
Applied to
→BP1 supports · met
Sources & reference links