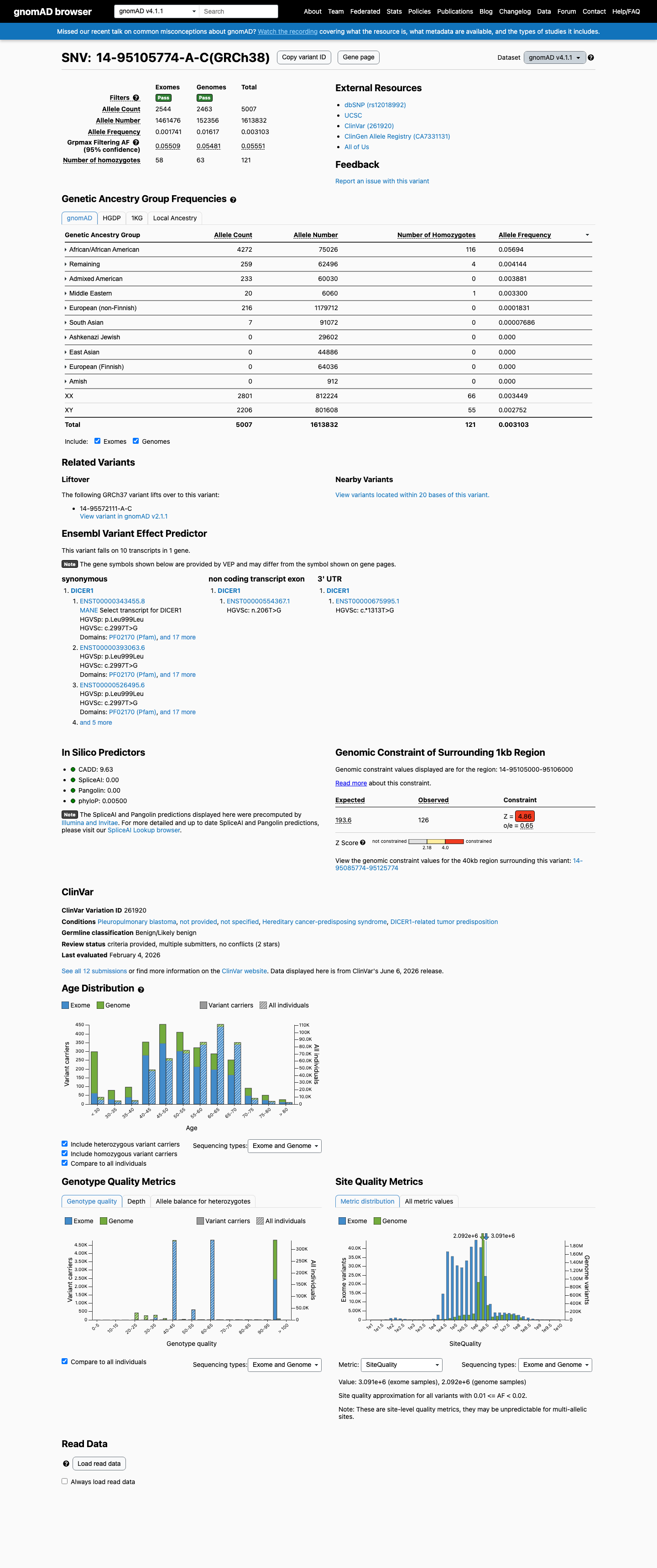
NM_177438.3:c.2997T>G (p.Leu999=) is a synonymous variant in exon 19 of DICER1. It is present at high frequency in gnomAD v4.1 with an overall allele frequency of 0.31% (5007/1613832 alleles) and 121 homozygotes. The highest subpopulation frequency is 5.69% in African/African American individuals (4272/75026 alleles, 116 homozygotes), exceeding the VCEP BA1 stand-alone benign threshold of >0.3%.1 BA1 (Stand-Alone Benign) is met: the African/African American subpopulation allele frequency of 5.69% exceeds the 0.3% threshold with >2000 alleles tested and ≥5 alleles present. A frequency of this magnitude is incompatible with a highly penetrant autosomal dominant tumor predisposition disorder.2 BS1 (Strong Benign) is independently met at the >0.03% threshold in the same subpopulation, subsumed by the stronger BA1 evidence.3 BS2_Supporting is met: 121 homozygous individuals are reported in gnomAD v4.1, satisfying the VCEP threshold of ≥2 homozygotes in individuals lacking clinical information.4 BP4_Supporting is met: SpliceAI predicts no splicing impact for this synonymous variant (max delta score = 0.03).5 BP7_Supporting is met: the variant is a synonymous change (p.Leu999=) and meets BP4, satisfying the DICER1 VCEP BP7 requirements.6 This variant has been classified as Benign by 10 clinical laboratories in ClinVar (VariationID: 261920), consistent with the population frequency evidence.7 No publications among the seven reviewed (PMID:25741868, 26467025, 24493721, 29474644, 24761742, 25394175, 28492532) specifically mention NM_177438.3:c.2997T>G or p.Leu999=. All are methodology, guideline, or gene-level review papers without variant-specific evidence.8 Under the Tavtigian point-based system (DICER1 VCEP v1.4.0): BA1 = -8 points. BS2_Supporting = -1 point. BP4_Supporting = -1 point. BP7_Supporting = -1 point. Total = -11 points. Classification: Benign (≤ -7).9
DICER1
Final classification
Benign
DICER1 c.2997T>G · p.Leu999=
DICER1
NM_177438.3:c.2997T>G (p.Leu999=) is a synonymous variant in exon 19 of DICER1. It is present at high frequency in gnomAD v4.1 with an overall allele frequency of 0.31% (5007/1613832 alleles) and 121 homozygotes. The highest subpopulation frequency is 5.69% in African/African American individuals (4272/75026 alleles, 116 homozygotes), exceeding the VCEP BA1 stand-alone benign threshold of >0.3%.
ClinGen DICER1 and miRNA-Processing Gene Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for DICER1 Version 1.4.0 v1.4.0 point-based framework: BA1 stand-alone benign (-8) + BS1 strong benign (-4) + BS2 supporting benign (-1) + BP4 supporting benign (-1) + BP7 supporting benign (-1) = -15 points, which maps to Benign.
Classification rationale
BA1BS1BS2BP4BP7
Benign
DICER1 c.2997T>G
BA1 + BS1 + BS2 + BP4 + BP7
→
Benign
Gene diagram
· NM_177438.3 · variants mapped to exon structure
DICER1
NM_177438.3
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 8 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
BA1
stand-alone
Benign
NM_177438.3:c.2997T>G has an allele frequency of 5.69% in the African/African American population in gnomAD v4.1 (4272/75026 alleles, 116 homozygotes), exceeding the DICER1 VCEP BA1 threshold of >0.3% in a subpopulation with >2000 alleles and ≥5 alleles present. This frequency is incompatible with a highly penetrant autosomal dominant tumor predisposition syndrome.
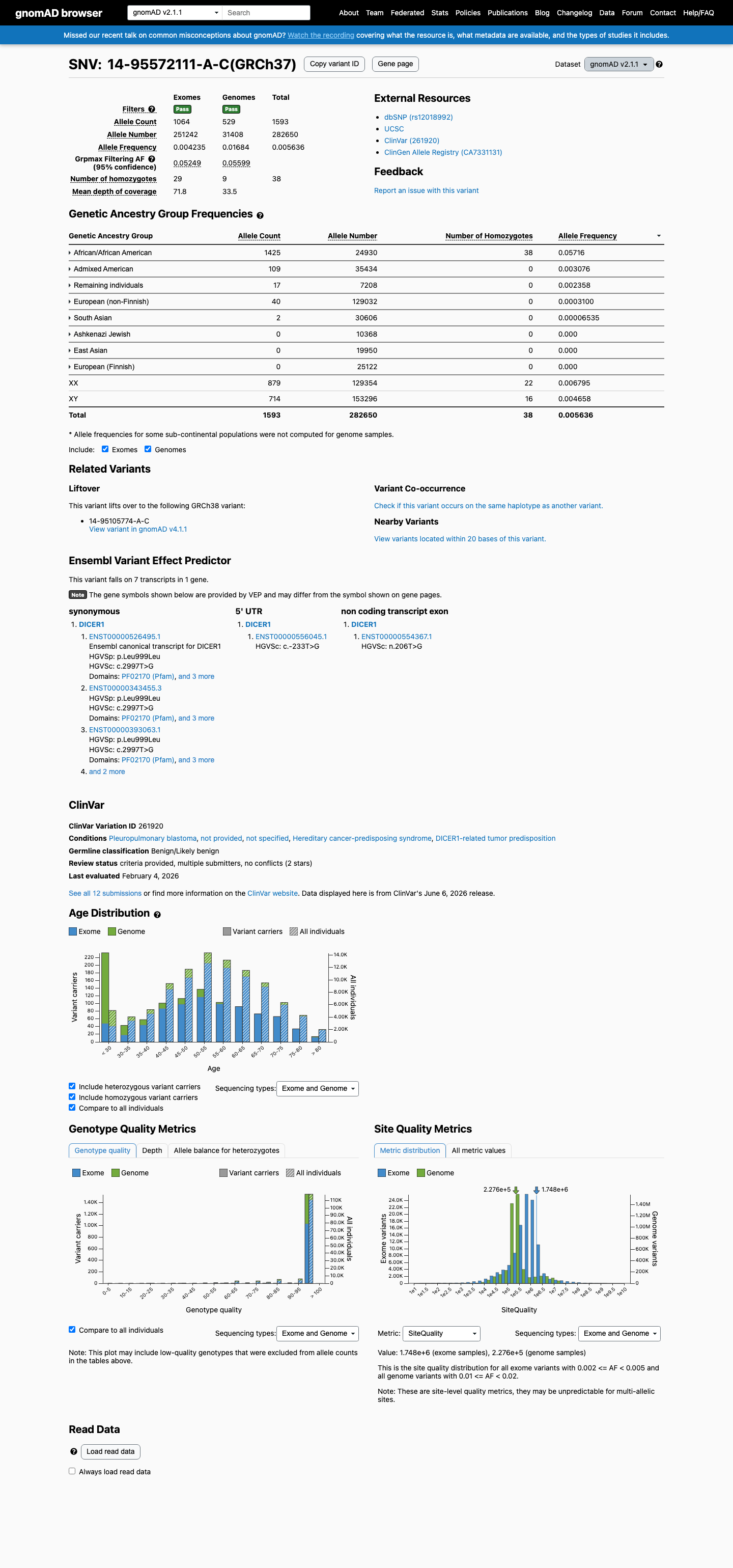
gnomAD v4.1 African/African American AF = 5.69% (4272/75026 alleles116 homozygotes)gnomAD v2.1 African/African American AF = 5.72% (1425/24930 alleles
✓
BS1
strong
Benign
The African/African American subpopulation AF of 5.69% in gnomAD v4.1 exceeds the DICER1 VCEP BS1 threshold of >0.03% with >2000 alleles and ≥5 alleles present. This criterion is independently met but subsumed by BA1.
gnomAD v4.1 African/African American AF = 5.69% exceeds BS1 threshold of 0.03%.
✓
BS2
supporting
Benign
Per DICER1 VCEP BS2_Supporting: '2+ observations of homozygosity in individuals lacking clinical information.' gnomAD v4.1 reports 121 homozygotes for NM_177438.3:c.2997T>G. The presence of numerous homozygous individuals in a population database is inconsistent with a pathogenic dominant tumor predisposition variant.
gnomAD v4.1: 121 homozygotesgnomAD v2.1: 38 homozygotes.
✓
BP4
supporting
Benign
Per DICER1 VCEP: for synonymous variants, concordance of MaxEntScan and SpliceAI showing no splicing effects qualifies for BP4_Supporting. SpliceAI predicts no splicing impact (max delta score = 0.03). This synonymous change at c.2997T>G is not predicted to create a cryptic splice site or disrupt splicing.
SpliceAI max delta = 0.03no predicted splicing alteration for this synonymous variant.
✓
BP7
supporting
Benign
NM_177438.3:c.2997T>G is a synonymous (silent) variant (p.Leu999=). Per DICER1 VCEP BP7_Supporting: 'Silent variant... Caveat: Variant must meet BP4 to apply BP7.' BP4 is met (SpliceAI predicts no splicing impact, delta = 0.03). This variant does not alter the amino acid sequence and is not predicted to affect splicing.
Synonymous variant p.(Leu999=)SpliceAI delta = 0.03 (no splicing effect)BP4 met.
Assessed · not applied
Pathogenic
PS2
No de novo observations identified for NM_177438.3:c.2997T>G in any reviewed publication or ClinVar submission.
PS3
No well-established in vitro or in vivo functional studies (RNA splicing assay or cleavage assay) have been performed for this specific variant.
PM2
Per DICER1 VCEP: PM2_Supporting requires allele frequency <0.000005 across gnomAD.
PP3
Per DICER1 VCEP: PP3_Supporting requires REVEL ≥ 0.750 for missense variants OR agreement in splicing predictors predicting splicing effects.
PP4
No somatic tumor testing data available demonstrating a DICER1 somatic hotspot second hit (at p.S1344, p.E1705, p.D1709, p.D1713, p.G1809, p.D1810, or p.E1813) with retention of the germline variant.
Benign
BS3
Per DICER1 VCEP BS3_Strong: requires 'no splicing impact observed via RNA assay (observed more than once).' No RNA assay data is available for this variant.
BS4
No segregation data available.
BP2
No observations of NM_177438.3:c.2997T>G in trans with a pathogenic/likely pathogenic DICER1 variant, nor in cis with 2+ different P/LP DICER1 variants, in any reviewed data source.
N/A · 12
PVS1 · PS1 · PS4 · PM1 · PM5 · PM6 · PP1 · PP2 · PP5 · BP1 · BP5 · BP6
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.00310255; MAF= 0.31026%, 5007/1613832 alleles, homozygotes = 121) and has highest observed frequency in the African/African American population (AF= 0.0569403; MAF= 5.69403%, 4272/75026 alleles, homozygotes = 116); grpmax FAF= 0.0555142.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.00563595; MAF= 0.56359%, 1593/282650 alleles, homozygotes = 38) and has highest observed frequency in the African/African American population (AF= 0.05716; MAF= 5.71600%, 1425/24930 alleles, homozygotes = 38); grpmax FAF= 0.0559887.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0036377456835704203, 67/18418 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.31%
· 5007 / 1,613,832
121 hom · FAF 5.6%
121 hom · FAF 5.6%
African/African American 4272 / 75,026 |
5.7% 116 hom |
Remaining individuals 259 / 62,496 |
0.41% 4 hom |
Admixed American 233 / 60,030 |
0.39% |
Middle Eastern 20 / 6,060 |
0.33% 1 hom |
European (non-Finnish) 216 / 1,179,712 |
0.018% |
South Asian 7 / 91,072 |
0.0077% |
+ 4 not observed (European (Finnish), Amish, East Asian, Ashkenazi Jewish)
gnomAD v2.1
0.56%
· 1593 / 282,650
38 hom · FAF 5.6%
38 hom · FAF 5.6%
African/African American 1425 / 24,930 |
5.7% 38 hom |
Admixed American 109 / 35,434 |
0.31% |
Remaining individuals 17 / 7,208 |
0.24% |
European (non-Finnish) 40 / 129,032 |
0.031% |
South Asian 2 / 30,606 |
0.0065% |
+ 3 not observed (Ashkenazi Jewish, East Asian, European (Finnish))
gnomAD Canada 🇨🇦
0.36%
· 67 / 18,418
0 hom · FAF 4.3%
0 hom · FAF 4.3%
African/African American 55 / 1,020 |
5.4% |
Middle Eastern 1 / 144 |
0.69% |
Latino/Admixed American 5 / 838 |
0.6% |
Remaining individuals 5 / 1,138 |
0.44% |
European (non-Finnish) 1 / 11,738 |
0.0085% |
+ 4 not observed (Ashkenazi Jewish, East Asian, European (Finnish), South Asian)
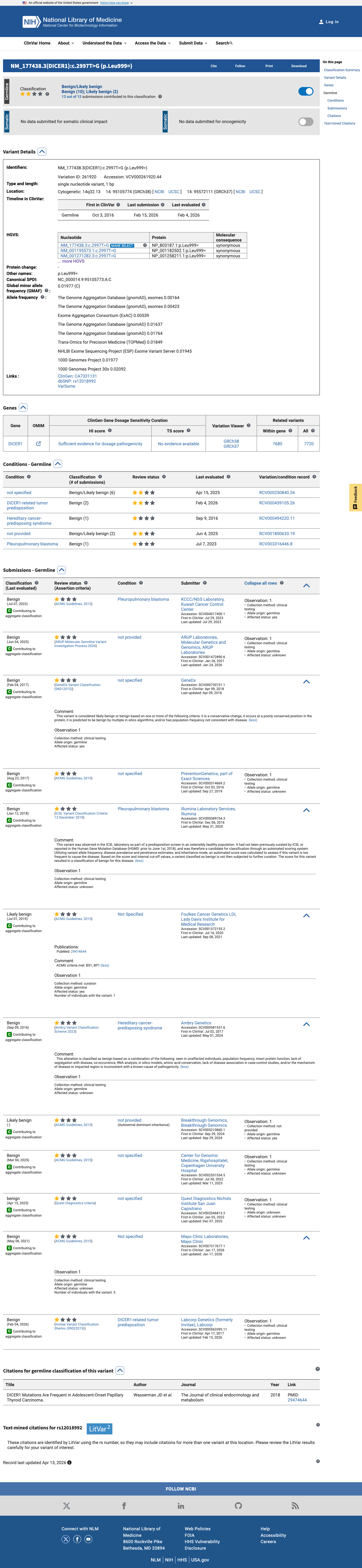
ClinVar
This variant has been reported in ClinVar as Benign (9 clinical laboratories) and as benign (1 clinical laboratory). (ClinVarID = 261920)
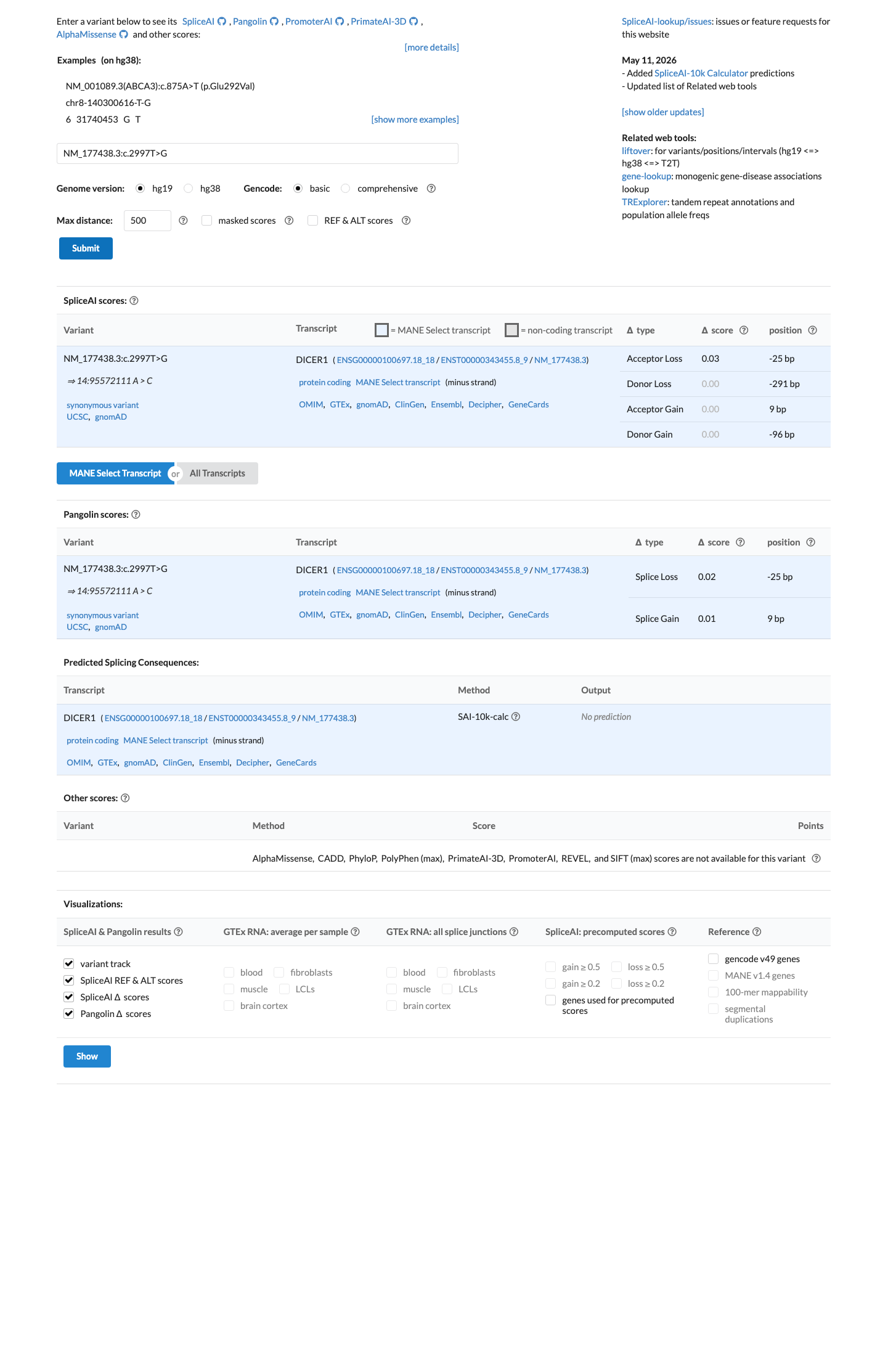
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.03).
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
9Sources
Triaged references · 7 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
24493721 ↗
American Society of Clinical Oncology Expert Statement: collection and use of a cancer family history for oncology providers.
CLINVAR
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR