NM_198253.2:c.833C>T (p.Pro278Leu) in TERT is a rare missense variant with extremely low population frequency (gnomAD v2.1 AF=9.83e-6, v4.1 AF=3.77e-6), meeting PM2 at supporting strength.1 Multiple in silico predictors (REVEL 0.137, BayesDel -0.245, SpliceAI max delta 0.02) consistently suggest no damaging effect on the gene product, meeting BP4 at supporting benign strength.2 This variant has been reported in ClinVar as Likely benign by a single clinical laboratory (Labcorp Genetics/Invitae, 1-star review status, SCV000824613). The classification and review status do not meet thresholds for PP5 or BP6 application under the 3-star expert panel requirement.3 No variant-specific functional data, de novo observations, segregation data, case-control studies, or domain-level hotspot evidence were identified. OncoKB reports Unknown Oncogenic Effect. PM1 is not met as Pro278 is not in a statistically significant mutational hotspot.4 PVS1 is not applicable (missense variant outside null-variant buckets). PS1, PM5, BP7, BP3, PM3, and PM4 are not applicable due to variant type or absence of required evidence patterns.5 With one supporting pathogenic criterion (PM2) and one supporting benign criterion (BP4), the evidence is in equipoise. The final classification under generic ACMG/AMP 2015 rules (Richards et al., PMID:25741868) is Variant of Uncertain Significance (VUS).6
TERT
Final classification
VUS
TERT c.833C>T · p.Pro278Leu
TERT
NM_198253.2:c.833C>T (p.Pro278Leu) in TERT is a rare missense variant with extremely low population frequency (gnomAD v2.1 AF=9.83e-6, v4.1 AF=3.77e-6), meeting PM2 at supporting strength.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting benign; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP4
VUS
TERT c.833C>T
PM2 + BP4
→
VUS
2
revelbayesdelspliceai ↗
4
oncokb ↗
5
pvs1_generic_framework ↗pm5_candidates
6
generic_acmg_combination_rules
Gene diagram
· NM_198253.2 · variants mapped to exon structure
TERT
NM_198253.2
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 20 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is extremely rare in population databases: gnomAD v2.1 allele frequency is 9.83e-6 (0.00098%, 2/203,400 alleles), v4.1 is 3.77e-6 (0.00038%, 6/1,590,960 alleles), and absent from gnomAD-Canada. All frequencies are well below the 0.1% PM2 threshold for non-VCEP frameworks.
gnomAD v2.1: AF=9.83e-6 (2/203400)highest subpopulation NFE AF=2.26e-5 (0.00226%)
✓
BP4
supporting
Benign
Multiple lines of computational evidence support no impact on the gene product: REVEL score 0.137 (below pathogenic threshold), BayesDel score -0.244664 (predicts benign effect), and SpliceAI max delta 0.02 (no predicted splicing alteration).
REVEL: 0.137 (not predicted damaging)BayesDel: -0.244664 (predicted benign)SpliceAI: max delta 0.02
Assessed · not applied
Pathogenic
PS1
No evidence of a different nucleotide change at codon 278 producing the same Pro278Leu missense change with established pathogenicity; PS1 requires a proven pathogenic same-amino-acid change via a different nucleotide variant.
PS2
No de novo occurrence data available for this variant in any publication or database.
PS3
No variant-specific functional studies identified.
PS4
No case-control data or statistically significant enrichment of this variant in affected individuals versus controls available.
PM1
Pro278 is not located within a statistically significant mutational hotspot (cancerhotspots.org negative).
PM6
No de novo occurrence reported for this variant in any publication or database.
PP1
No segregation data available for this variant in affected families.
PP2
Insufficient data to establish that TERT has a low rate of benign missense variation.
PP3
Multiple in silico predictors do not support a damaging effect: REVEL score is 0.137 (well below ~0.5 pathogenic threshold), BayesDel is -0.245 (negative score consistent with benign effect), and SpliceAI predicts no splicing impact (max delta 0.02).
PP4
No patient phenotype or family history data specific to this variant are available to assess whether the clinical presentation matches TERT-related telomere biology disorders.
PP5
ClinVar classification is Likely benign (not pathogenic) from a single submitter (Labcorp Genetics/Invitae).
Benign
BA1
gnomAD allele frequencies (v2.1: 0.00098%, v4.1: 0.00038%) are well below the 1% BA1 threshold for non-VCEP frameworks.
BS1
gnomAD allele frequencies (v2.1: 0.00098%, v4.1: 0.00038%) are well below the 0.3% BS1 threshold.
BS2
No homozygous observations in gnomAD and no evidence that this variant has been observed in healthy adults in the context of a fully penetrant dominant disorder.
BS3
No well-established in vitro or in vivo functional studies demonstrate no damaging effect for this variant.
BS4
No segregation data demonstrating lack of cosegregation with disease in affected families.
BP1
TERT-related disease is known to be caused by both missense and truncating variants.
BP2
No observation of this variant in trans with a known pathogenic variant for a fully penetrant dominant disorder.
BP5
No observation of this variant in a case where an alternative molecular basis for disease has been identified.
BP6
ClinVar review status is 1-star (criteria provided, single submitter).
N/A · 6
PVS1 · PM3 · PM4 · PM5 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
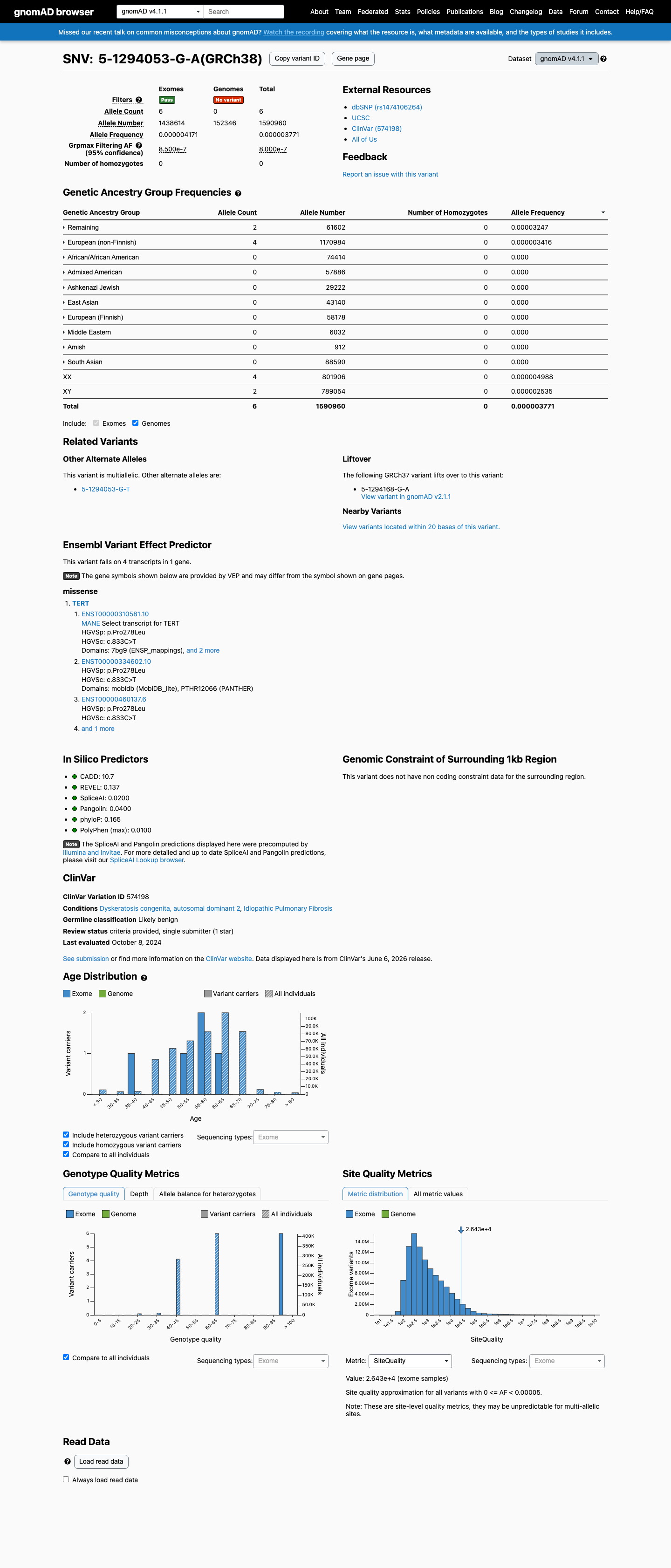
This variant is present in gnomAD v4.1 (AF= 3.77131e-06; MAF= 0.00038%, 6/1590960 alleles, homozygotes = 0) and has highest observed frequency in the Remaining individuals population (AF= 3.24665e-05; MAF= 0.00325%, 2/61602 alleles, homozygotes = 0); grpmax FAF= 8e-07.
v2.1
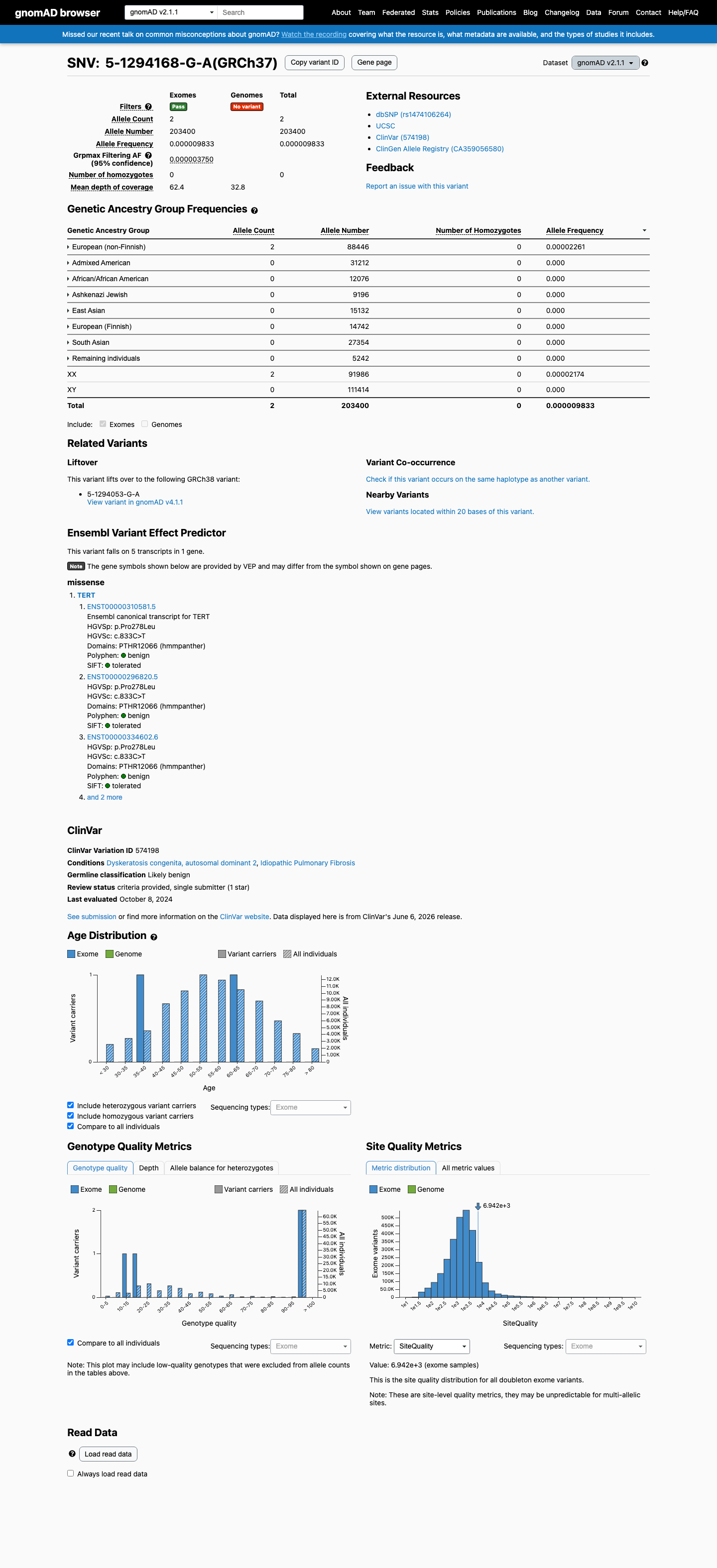
This variant is present in gnomAD v2.1 (AF= 9.83284e-06; MAF= 0.00098%, 2/203400 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 2.26127e-05; MAF= 0.00226%, 2/88446 alleles, homozygotes = 0); grpmax FAF= 3.75e-06.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00038%
· 6 / 1,590,960
0 hom · FAF 8e-05%
0 hom · FAF 8e-05%
Remaining individuals 2 / 61,602 |
0.0032% |
European (non-Finnish) 4 / 1,170,984 |
0.00034% |
+ 8 not observed (Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.00098%
· 2 / 203,400
0 hom · FAF 0.00038%
0 hom · FAF 0.00038%
European (non-Finnish) 2 / 88,446 |
0.0023% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
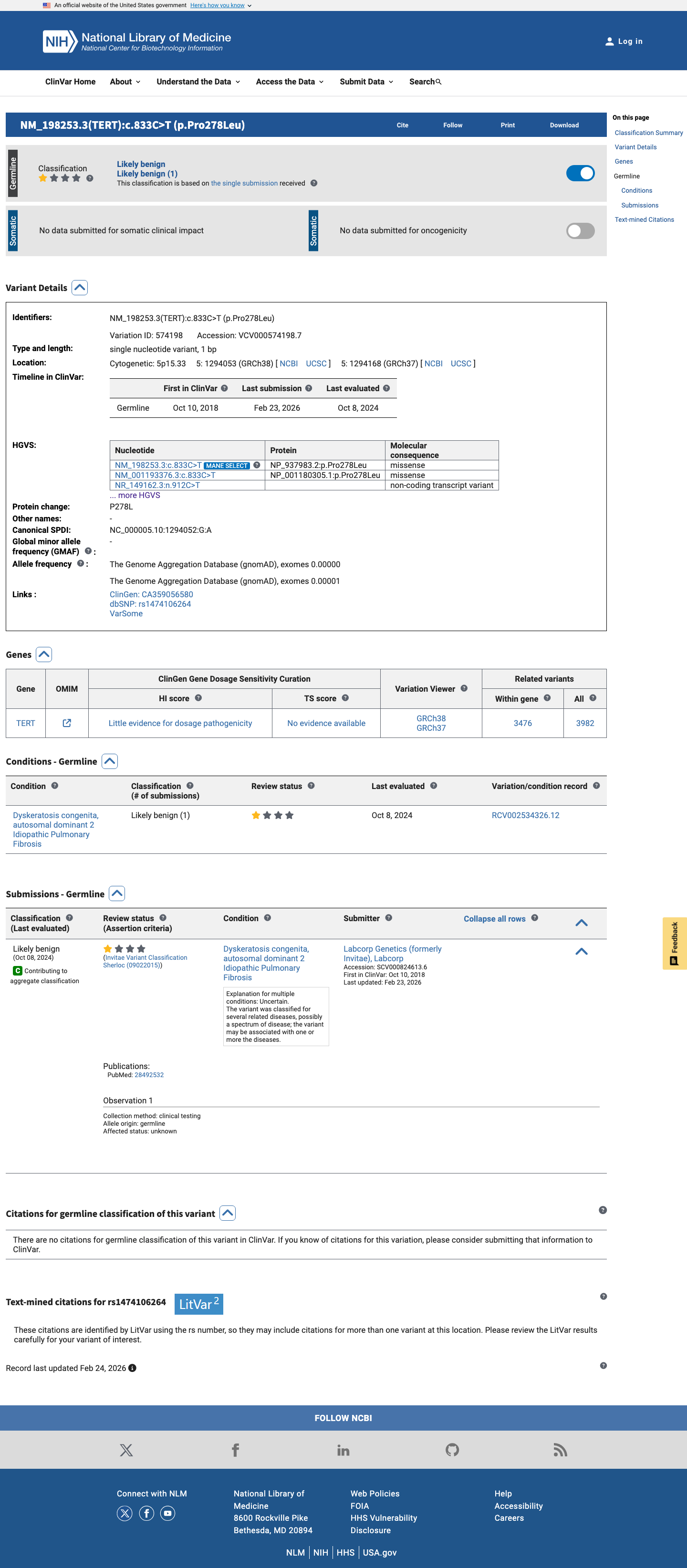
This variant has been reported in ClinVar as Likely benign (1 clinical laboratory). (ClinVarID = 574198)
In silico
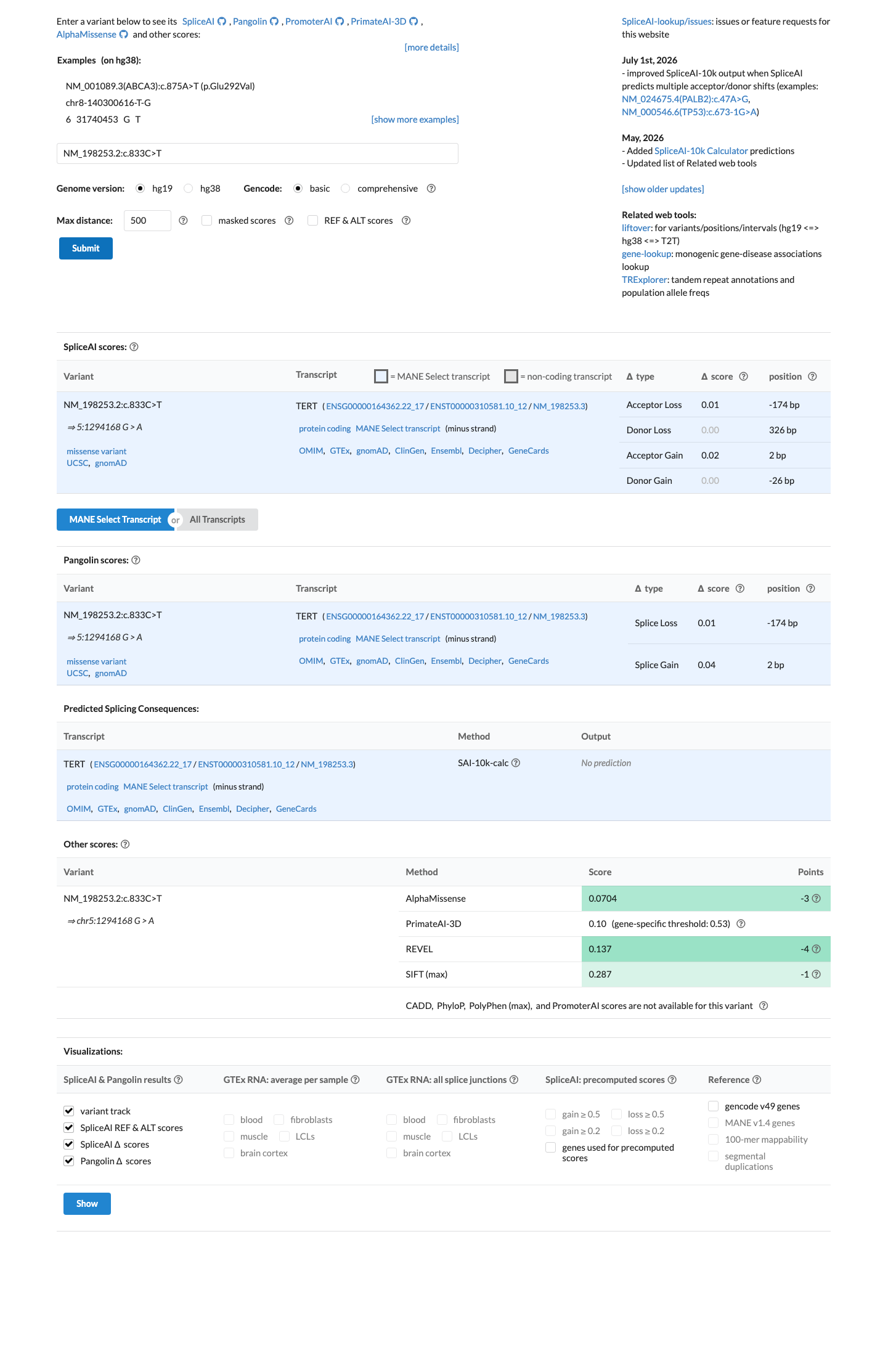
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02). REVEL score = 0.137. BayesDel score = -0.244664.
Functional
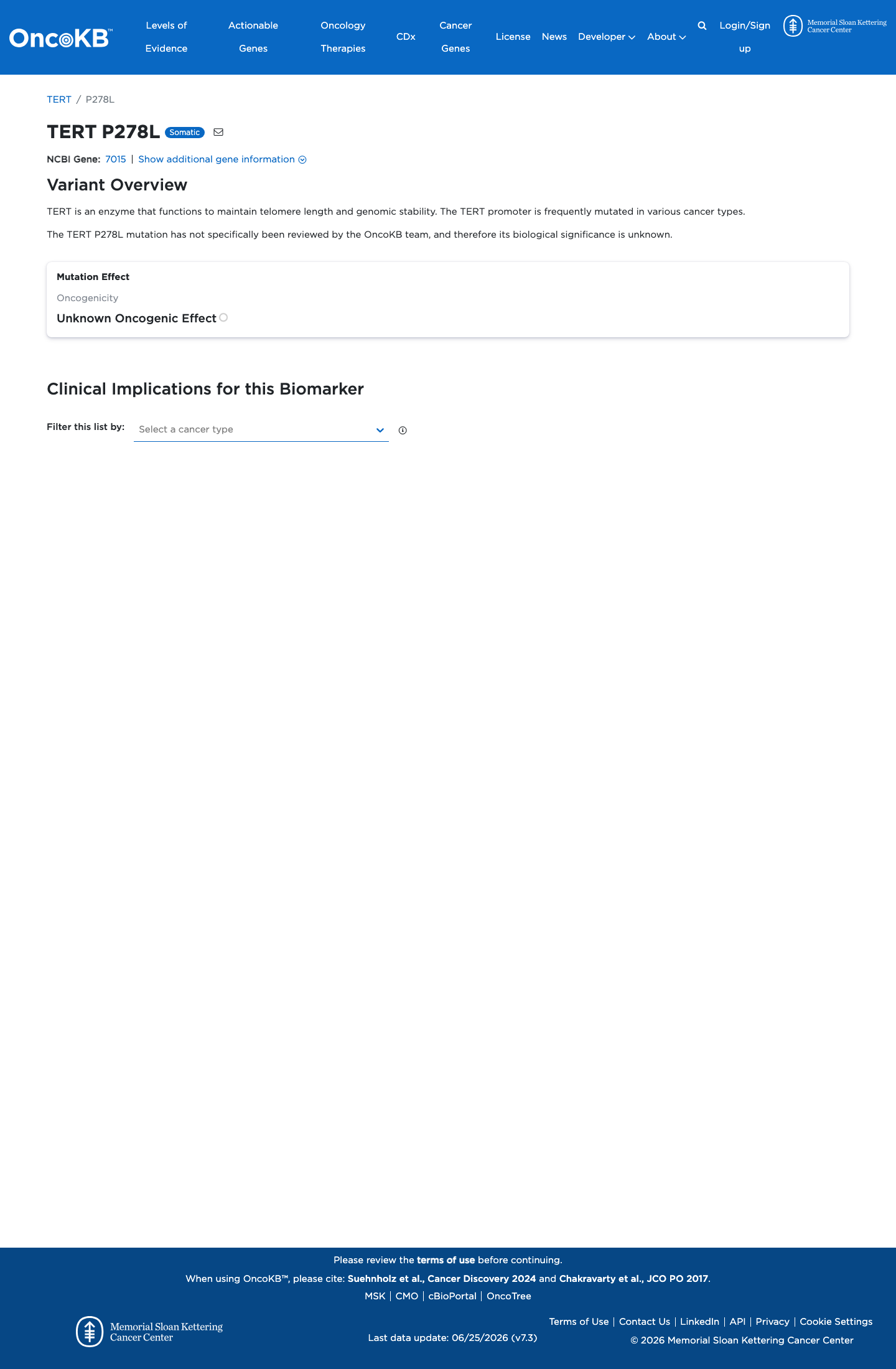
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. TERT is an enzyme that functions to maintain telomere length and genomic stability. The TERT promoter is frequently mutated in various cancer types.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 2 PMIDs not cited in assessment
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR