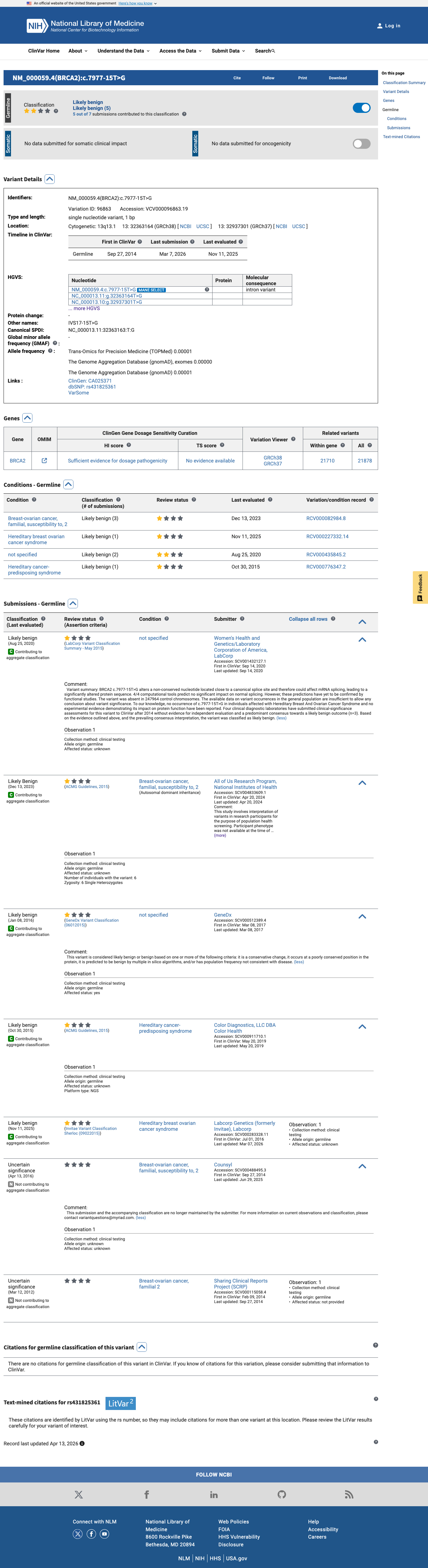
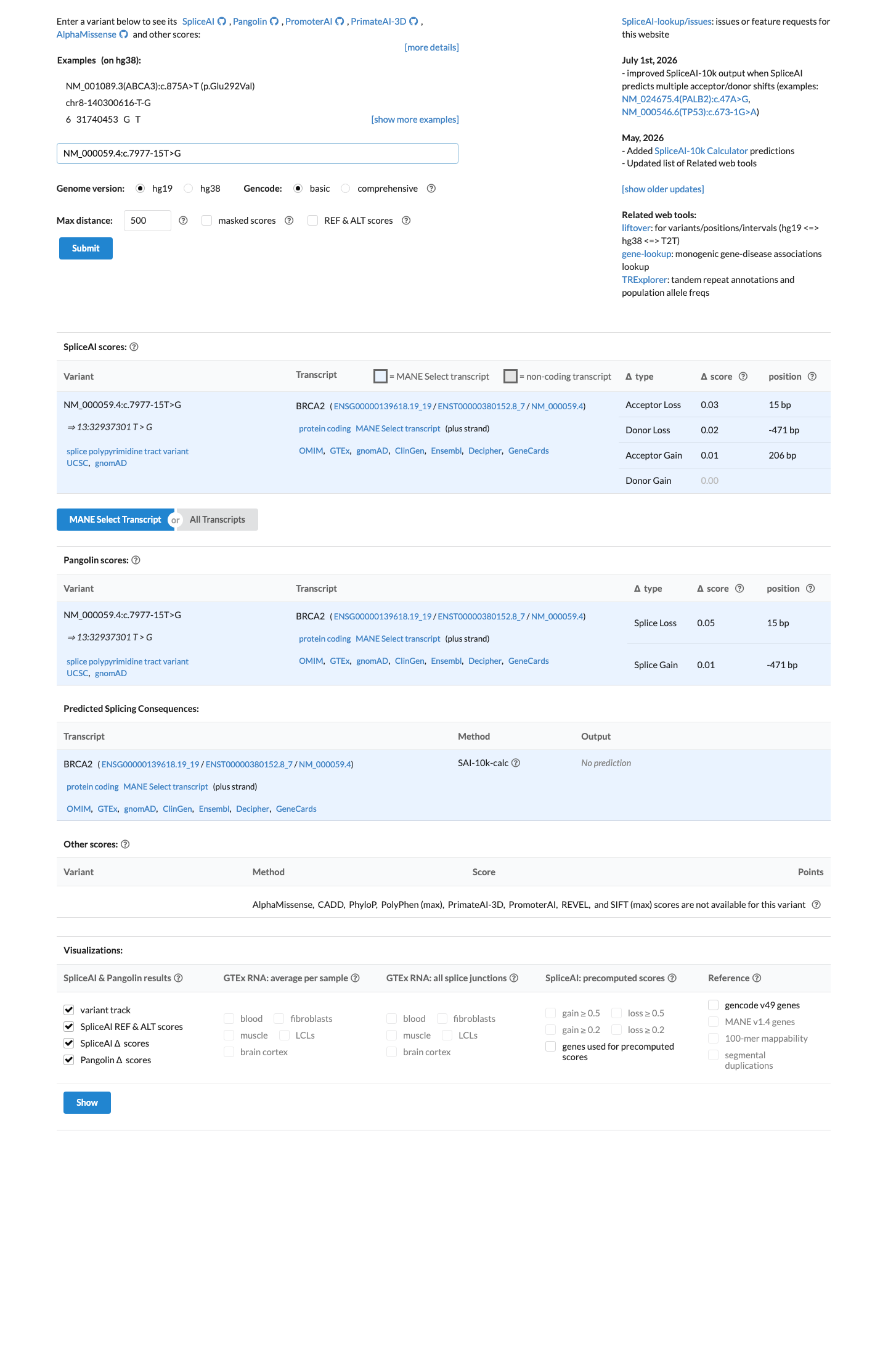
NM_000059.4:c.7977-15T>G is an intronic substitution located 15 bases upstream of BRCA2 exon 18, outside the canonical acceptor splice site consensus (+/-1,2). SpliceAI predicts no significant impact on splicing (max delta = 0.03), supporting a benign bioinformatic assessment under ENIGMA BP4_Supporting.1 This variant is extremely rare in population databases, present in 2 of 1,604,172 alleles in gnomAD v4.1 (AF = 1.25e-06), both in the European non-Finnish population. It is absent from gnomAD v2.1. The FAF of 2.8e-07 does not meet ENIGMA BS1 thresholds for benign population evidence, nor does it meet BA1 stand-alone benign criteria.2 In a multifactorial likelihood analysis by Parsons et al. 2019 (PMID:31131967), this variant had a combined likelihood ratio of 1.11 (co-occurrence LR = 1.02; family history LR = 1.08), which falls within the neutral zone and does not provide evidence for either pathogenicity (PP4) or benignity (BP5).3 The variant has been reported in ClinVar as Likely benign by 4 clinical laboratories and as Uncertain significance by 2 laboratories (ClinVar ID: 96863), though no expert panel classification has been assigned.4 The adjacent canonical splice acceptor variant c.7977-1G>C is a well-established pathogenic variant (Parsons et al. 2019 posterior probability = 0.999; IARC Class 5), but c.7977-15T>G does not disrupt the splice consensus and is predicted to be splicing-neutral by SpliceAI.5 Overall, only BP4_Supporting is met. Under the ENIGMA Table 3 combining rules, a single supporting benign criterion does not reach the threshold for Likely Benign (which requires either Strong_benign + Supporting_benign or >= 2 Supporting_benign). This variant is classified as a Variant of Uncertain Significance (VUS).6
BRCA2
Final classification
VUS
BRCA2 c.7977-15T>G · p.?
BRCA2
NM_000059.4:c.7977-15T>G is an intronic substitution located 15 bases upstream of BRCA2 exon 18, outside the canonical acceptor splice site consensus (+/-1,2). SpliceAI predicts no significant impact on splicing (max delta = 0.03), supporting a benign bioinformatic assessment under ENIGMA BP4_Supporting.
Only BP4_Supporting is met (SpliceAI max delta 0.03, intronic variant outside +/-1,2 acceptor splice consensus predicts no splicing impact). Under ENIGMA Table 3 combining rules, a single Supporting_benign criterion does not reach the threshold for Likely Benign (requires either 2+ Supporting_benign, or 1 Strong_benign + 1 Supporting_benign, or 1 Moderate_benign + 1 Supporting_benign). Under the ENIGMA point system for conflicting evidence, BP4_Supporting = -1 falls in the VUS range (-1 to 5). No pathogenic criteria are met.
Classification rationale
BP4
VUS
BRCA2 c.7977-15T>G
BP4
→
VUS
Gene diagram
· NM_000059.4 · variants mapped to exon structure
BRCA2
NM_000059.4
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 14 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
BP4
supporting
Benign
Intronic variant at c.7977-15, outside the native acceptor splice site +/-1,2 positions. SpliceAI max delta score = 0.03 (<= 0.1), predicting no significant splicing impact. Meets ENIGMA BP4_Supporting criteria for intronic variants outside canonical splice sites with no predicted splicing effect.
c.7977-15 is outside +/-12 acceptor splice consensus positions.SpliceAI max delta = 0.03 (DS_AG=0.01
Assessed · not applied
Pathogenic
PS1
No previously classified pathogenic or likely pathogenic variant identified at the c.7977-15 position with the same predicted splicing effect.
PS3
Not listed in ENIGMA Specifications Table 9 (curated functional assay results for BRCA2).
PS4
No case-control study data available.
PM2
Under ENIGMA PM2_Supporting, the variant must be absent from both gnomAD v2.1 (non-cancer, exome only) and gnomAD v3.1 (non-cancer).
PP1
No co-segregation data available.
PP3
ENIGMA PP3 for intronic variants outside donor/acceptor +/-1,2 positions requires SpliceAI delta >= 0.2.
PP4
Not found in the Li et al.
Benign
BA1
ENIGMA BA1 requires FAF > 0.1% (0.001) in gnomAD non-founder populations.
BS1
ENIGMA BS1_Strong requires FAF > 0.01% (0.0001); BS1_Supporting requires FAF > 0.002% (0.00002) and <= 0.01%.
BS2
ENIGMA BS2 requires assessment of absence of Fanconi Anemia phenotype features in probands with co-occurrent variants in BRCA2.
BS3
Not listed in ENIGMA Specifications Table 9 (curated functional assay results for BRCA2).
BS4
ENIGMA BS4 requires quantitative lack of segregation in affected family members (LR <= 0.48 for Supporting).
BP5
Not found in the Li et al.
BP7
ENIGMA BP7_Supporting for intronic variants requires position at or beyond +7 (donor) or -21 (acceptor) AND BP4 met.
N/A · 13
PVS1 · PS2 · PM1 · PM3 · PM4 · PM5 · PM6 · PP2 · PP5 · BP1 · BP2 · BP3 · BP6
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
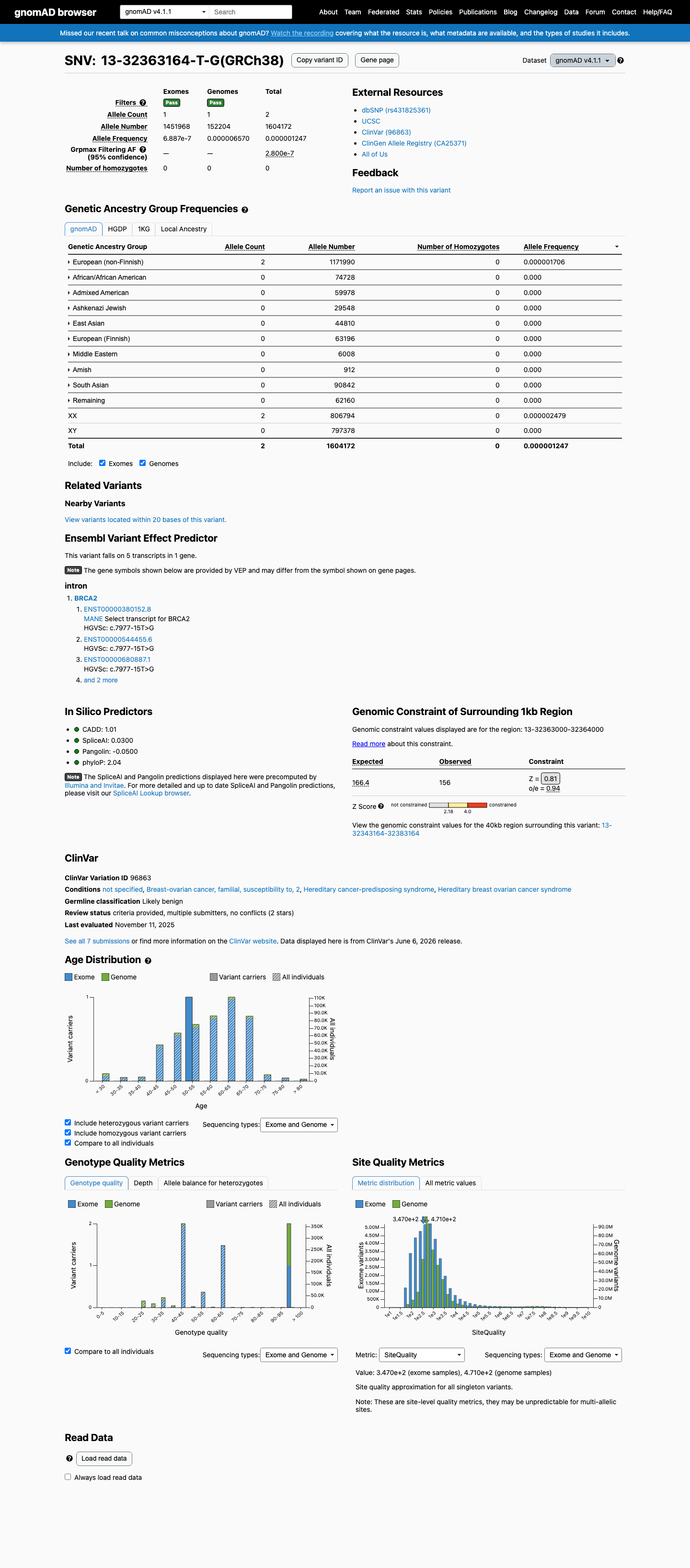
This variant is present in gnomAD v4.1 (AF= 1.24675e-06; MAF= 0.00012%, 2/1604172 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 1.7065e-06; MAF= 0.00017%, 2/1171990 alleles, homozygotes = 0); grpmax FAF= 2.8e-07.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00012%
· 2 / 1,604,172
0 hom · FAF 2.8e-05%
0 hom · FAF 2.8e-05%
European (non-Finnish) 2 / 1,171,990 |
0.00017% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Likely benign (4 clinical laboratories) and as Uncertain significance (2 clinical laboratories) and as Likely Benign (1 clinical laboratory). (ClinVarID = 96863)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.03).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 7 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
12692171 ↗
American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility.
CLINVAR
20065170 ↗
American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility.
CLINVAR
23188549 ↗
NSGC practice guideline: risk assessment and genetic counseling for hereditary breast and ovarian cancer.
CLINVAR
24493721 ↗
American Society of Clinical Oncology Expert Statement: collection and use of a cancer family history for oncology providers.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR