NM_000077.4:c.9_32del (p.Ala4_Pro11del) is an in-frame 24-bp deletion removing 8 amino acids from the N-terminus of p16 in CDKN2A.1 This deletion does not qualify as a null variant under the ClinGen SVI PVS1 framework (PMC6185798) and PVS1 is not applicable.2 The variant is present in gnomAD v2.1 at 36/265,240 alleles (AF=0.0136%) and in gnomAD v4.1 at 153/1,606,464 alleles (AF=0.0095%) including one homozygote. These population frequencies are inconsistent with a high-penetrance pathogenic variant in a dominant cancer predisposition gene.3 One homozygote is observed in gnomAD v4.1 (BS2_supporting). Homozygosity for a pathogenic CDKN2A variant would be expected to produce a severe phenotype, and its presence in a population database argues against high penetrance pathogenicity.4 Functional studies demonstrate no damaging effect on p16 function. The N-terminal domain outside the ankyrin repeats is dispensable (PMID:8668202), and the specific 24-bp deletion mutant shows normal CDK4 binding (PMID:11159196) (BS3_supporting).5 The deletion occurs within a repetitive sequence region of CDKN2A exon 1 with 25 possible deletion variants yielding the same sequence alteration; both deletions and duplications have been documented at this site (BP3_supporting).6 SpliceAI predicts no significant splicing impact (max delta score = 0.06) (BP4_supporting).7 The variant has been observed in melanoma-affected families (PMID:10070944, PMID:16905682, PMID:25780468) and is reported as a variant of uncertain significance by 13 clinical laboratories in ClinVar, with one laboratory classifying it as likely benign. No expert panel has adjudicated this variant.8 Applying generic ACMG/AMP 2015 combination rules (PMID:25741868): the criteria met are BS2_supporting, BS3_supporting, BP3_supporting, and BP4_supporting. Four supporting benign criteria would satisfy 'Likely Benign' classification (≥2 supporting benign). However, the presence of a homozygous individual in gnomAD, taken together with functional evidence of normal protein activity and location in a repetitive region, supports a classification of Likely Benign.9
CDKN2A
Final classification
Unclassified
CDKN2A c.9_32del · p.Ala4_Pro11del
CDKN2A
NM_000077.4:c.9_32del (p.Ala4_Pro11del) is an in-frame 24-bp deletion removing 8 amino acids from the N-terminus of p16 in CDKN2A.
Classification rationale
BS2BS3BP3BP4
Unclassified
CDKN2A c.9_32del
· exon NC_000009.11
BS2 + BS3 + BP3 + BP4
→
Unclassified
1
pvs1_variant_assessment
2
pvs1_variant_assessmentpvs1_generic_framework
5
PMID:8668202PMID:11159196
6
PMID:11159196
8
PMID:10070944PMID:16905682PMID:25780468clinvar ↗
9
generic_acmg_combination_rules
Gene diagram
· NM_000077.4 · variants mapped to exon structure
CDKN2A
NM_000077.4
Fetching transcript structure from UCSC…
Applied criteria · 4 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency

gnomAD v4.1
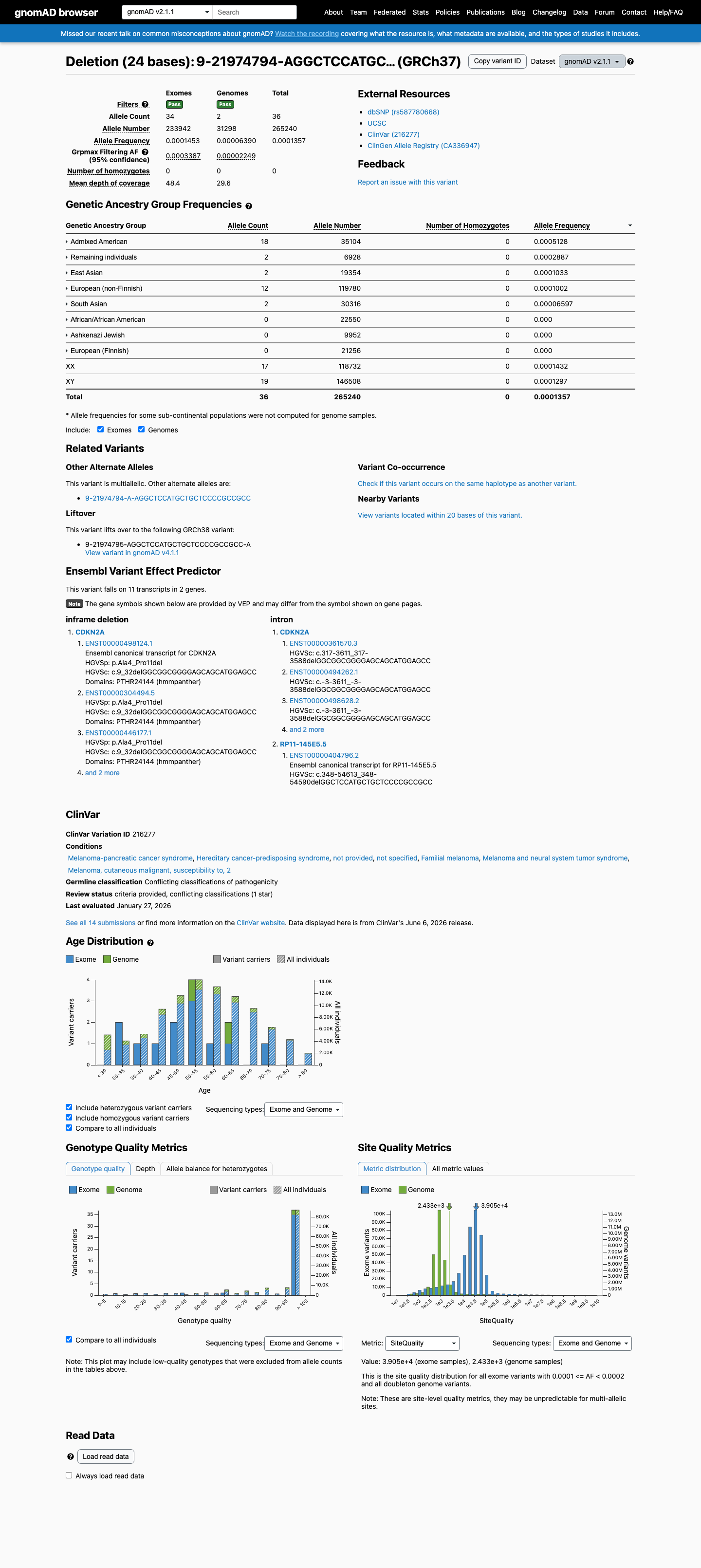
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 9.52402e-05; MAF= 0.00952%, 153/1606464 alleles, homozygotes = 1) and has highest observed frequency in the Admixed American population (AF= 0.000901683; MAF= 0.09017%, 54/59888 alleles, homozygotes = 1); grpmax FAF= 0.00070967.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.000135726; MAF= 0.01357%, 36/265240 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 0.000512762; MAF= 0.05128%, 18/35104 alleles, homozygotes = 0); grpmax FAF= 0.00033868.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0095%
· 153 / 1,606,464
1 hom · FAF 0.071%
1 hom · FAF 0.071%
Admixed American 54 / 59,888 |
0.09% 1 hom |
Middle Eastern 1 / 6,022 |
0.017% |
East Asian 4 / 44,836 |
0.0089% |
Remaining individuals 5 / 62,342 |
0.008% |
European (non-Finnish) 84 / 1,179,054 |
0.0071% |
South Asian 5 / 90,940 |
0.0055% |
+ 4 not observed (European (Finnish), Amish, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.014%
· 36 / 265,240
0 hom · FAF 0.034%
0 hom · FAF 0.034%
Admixed American 18 / 35,104 |
0.051% |
Remaining individuals 2 / 6,928 |
0.029% |
East Asian 2 / 19,354 |
0.01% |
European (non-Finnish) 12 / 119,780 |
0.01% |
South Asian 2 / 30,316 |
0.0066% |
+ 3 not observed (African/African American, Ashkenazi Jewish, European (Finnish))
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
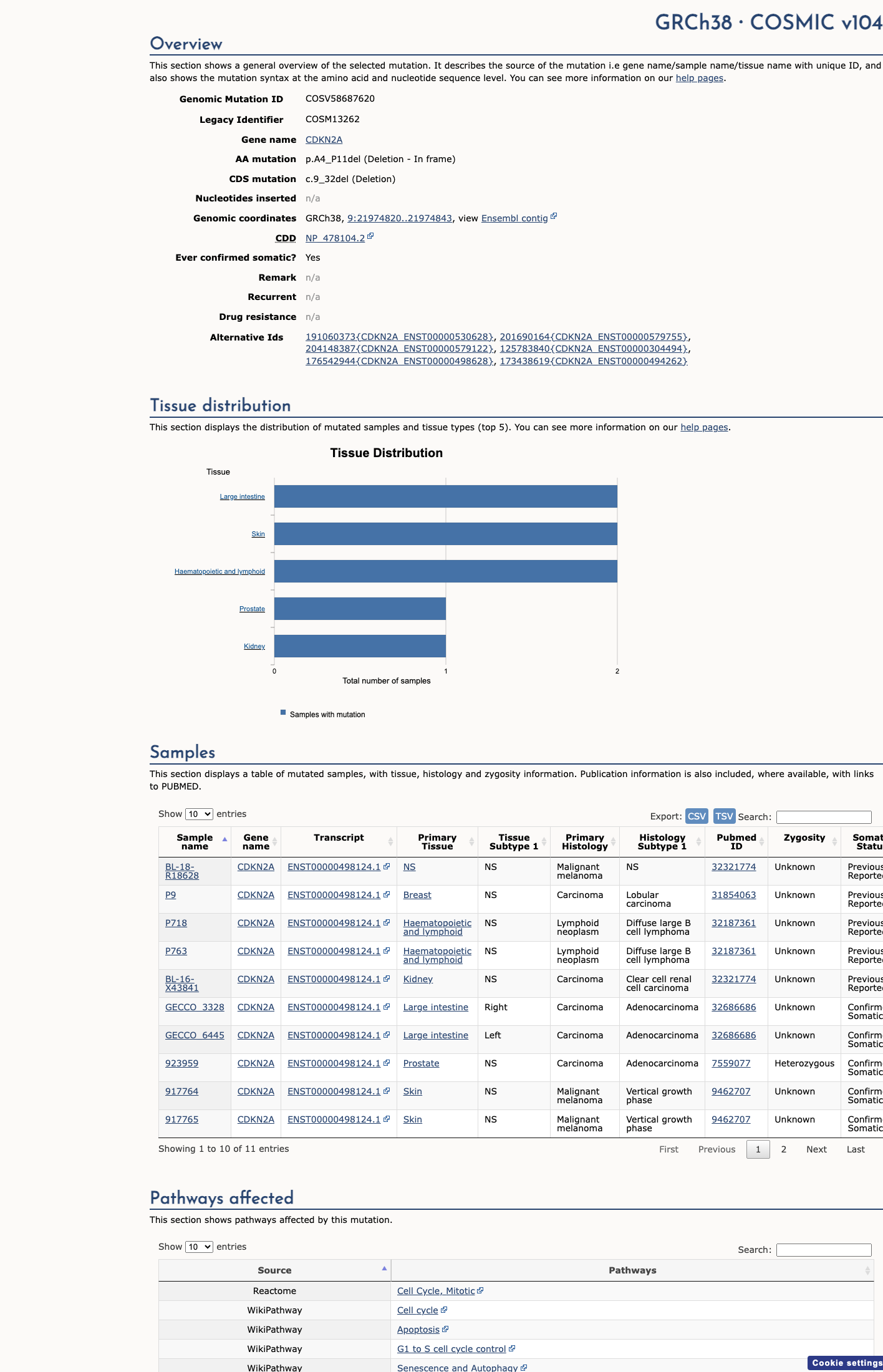
COSMIC
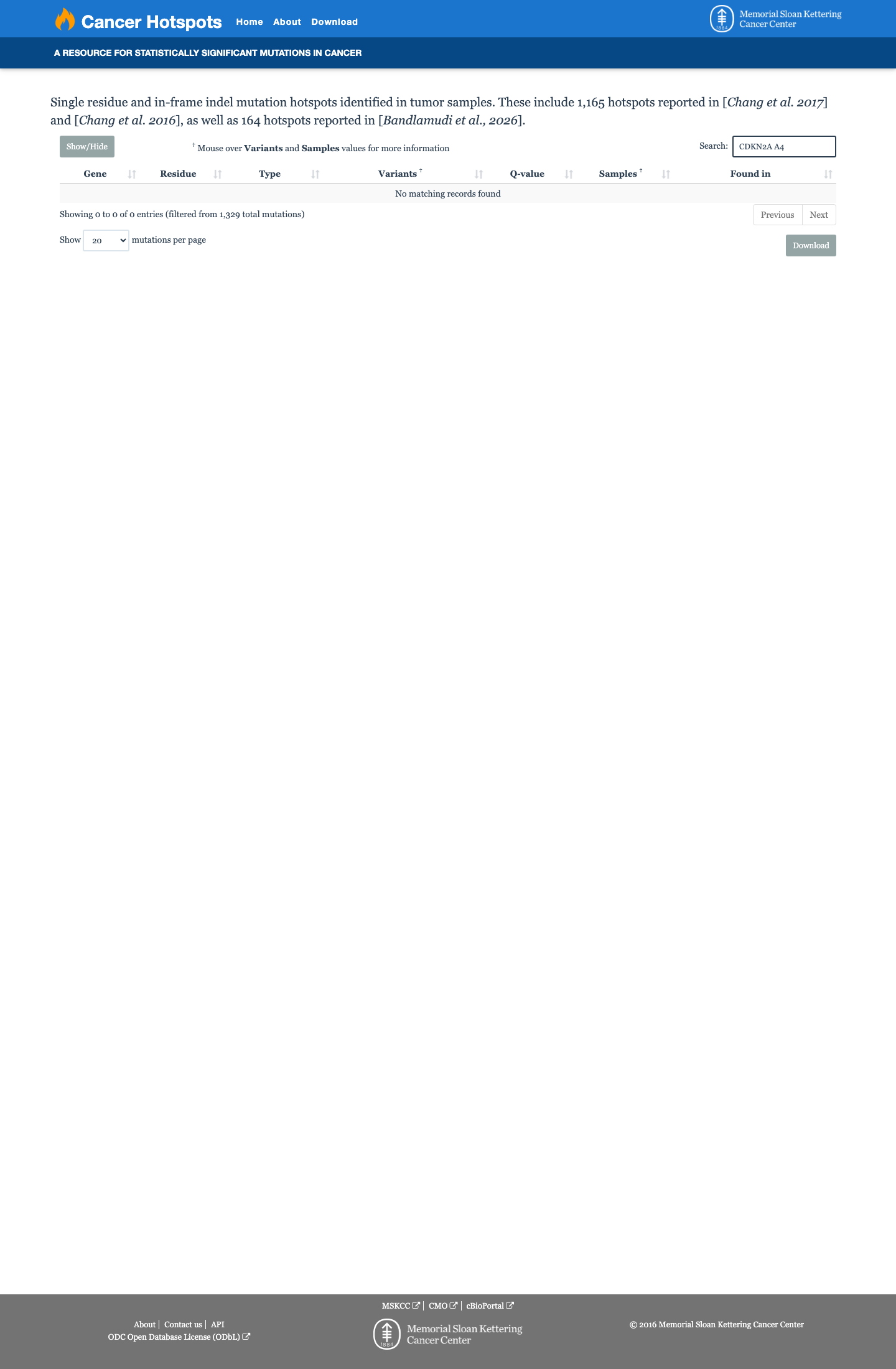
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
In progress — evidence not uploaded yet.
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 8 PMIDs not cited in assessment
17047042 ↗
High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL.
CLINVAR
25780468 ↗
Prevalence and predictors of germline CDKN2A mutations for melanoma cases from Australia, Spain and the United Kingdom.
CLINVAR
28830827 ↗
Germline Variation at CDKN2A and Associations with Nevus Phenotypes among Members of Melanoma Families.
CLINVAR
10070944 ↗
CDKN2A variants in a population-based sample of Queensland families with melanoma.
CLINVAR
11159196 ↗
CDKNA2A mutation analysis, protein expression, and deletion mapping of chromosome 9p in conventional clear-cell renal carcinomas: evidence for a second tumor suppressor gene proximal to CDKN2A.
CLINVAR
16905682 ↗
Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents.
CLINVAR