Classification rationale
BS1BP7
Likely Benign
MEN1 c.1311G>A
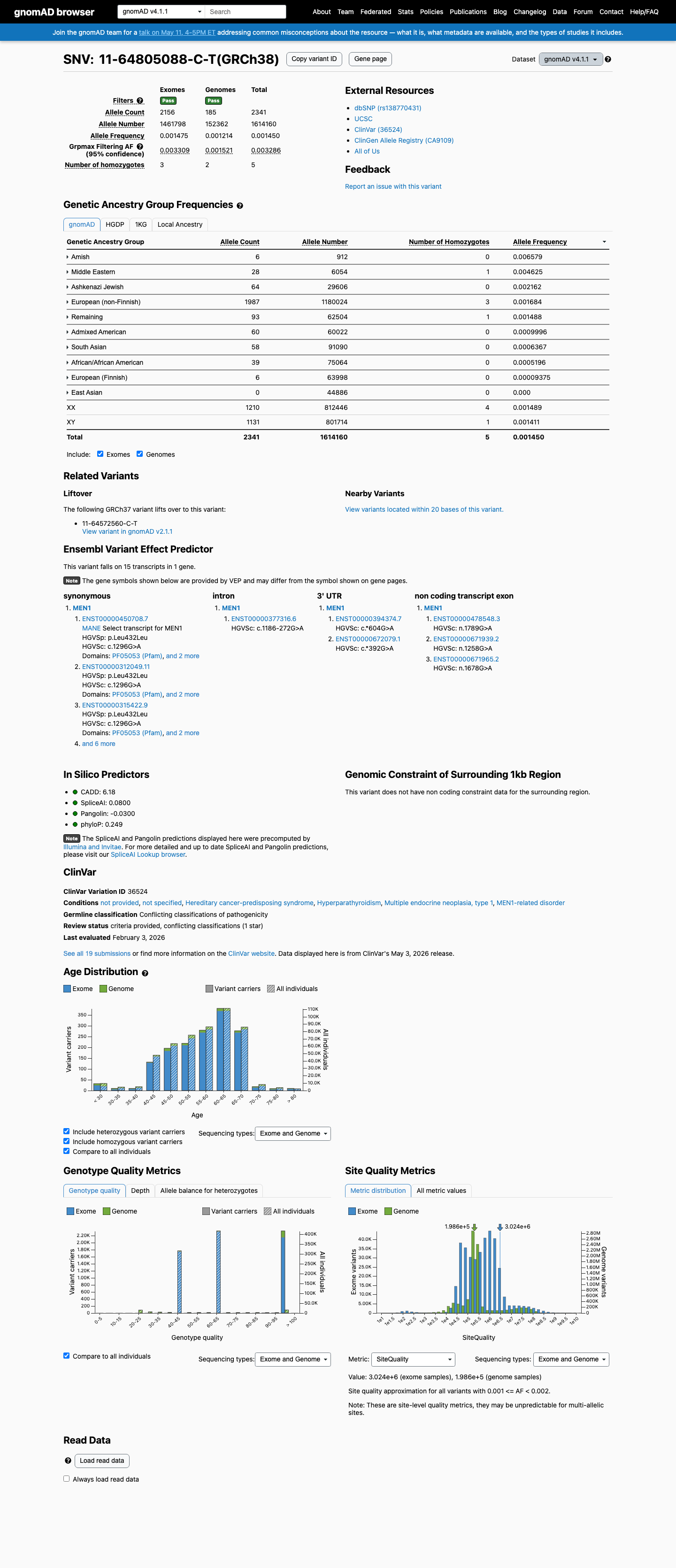
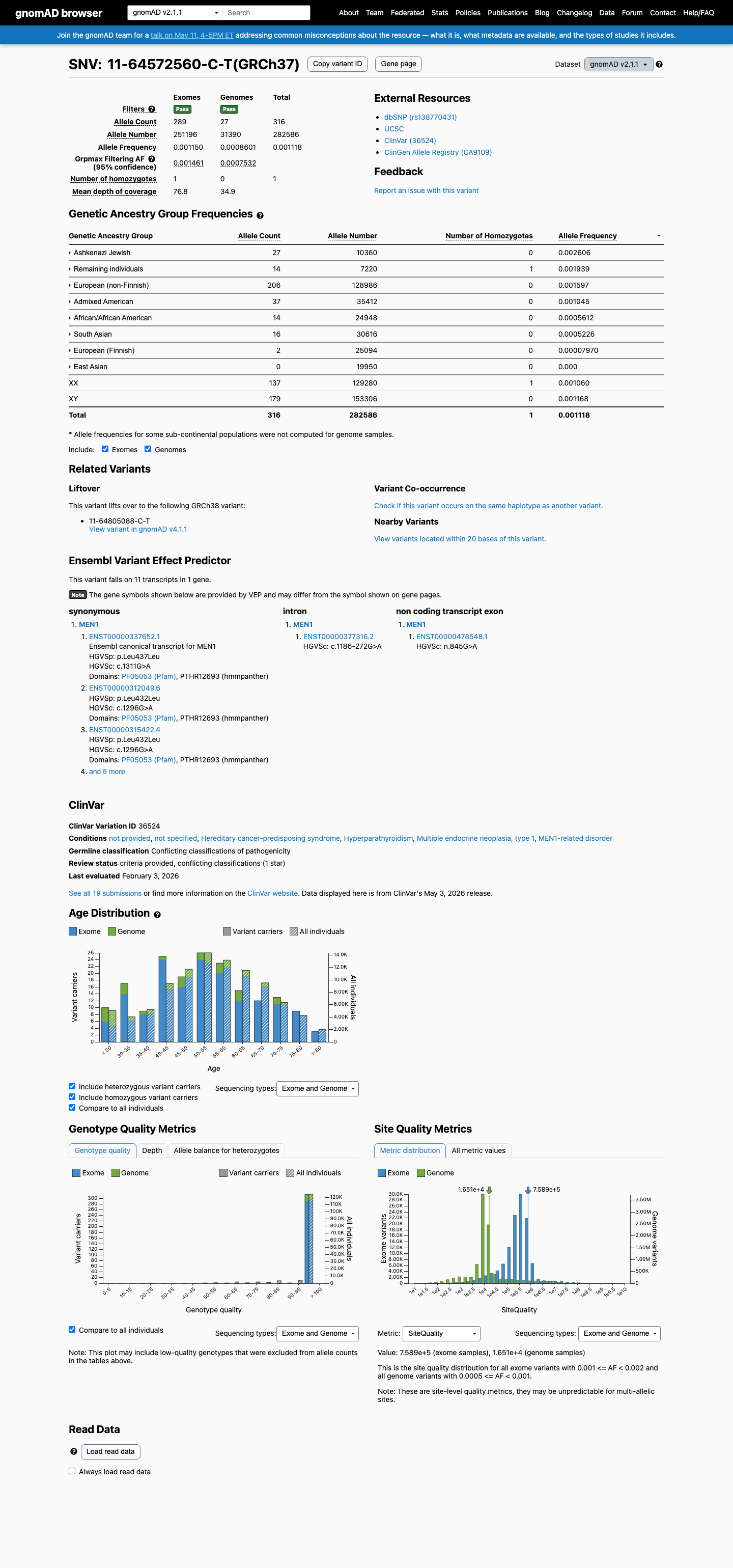
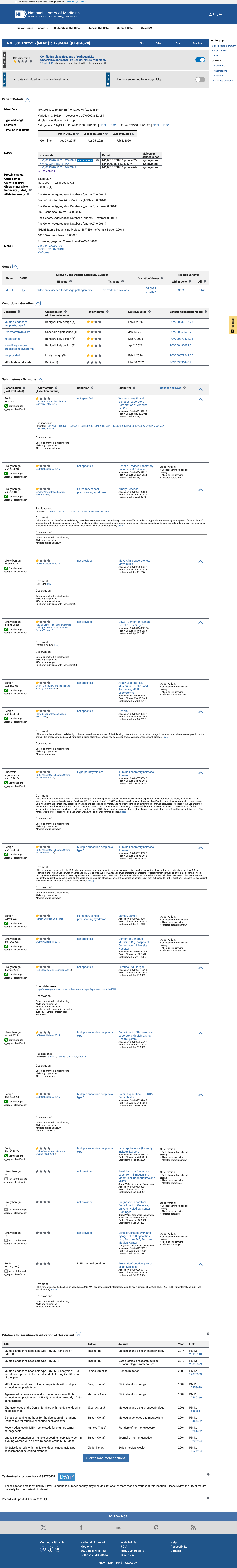
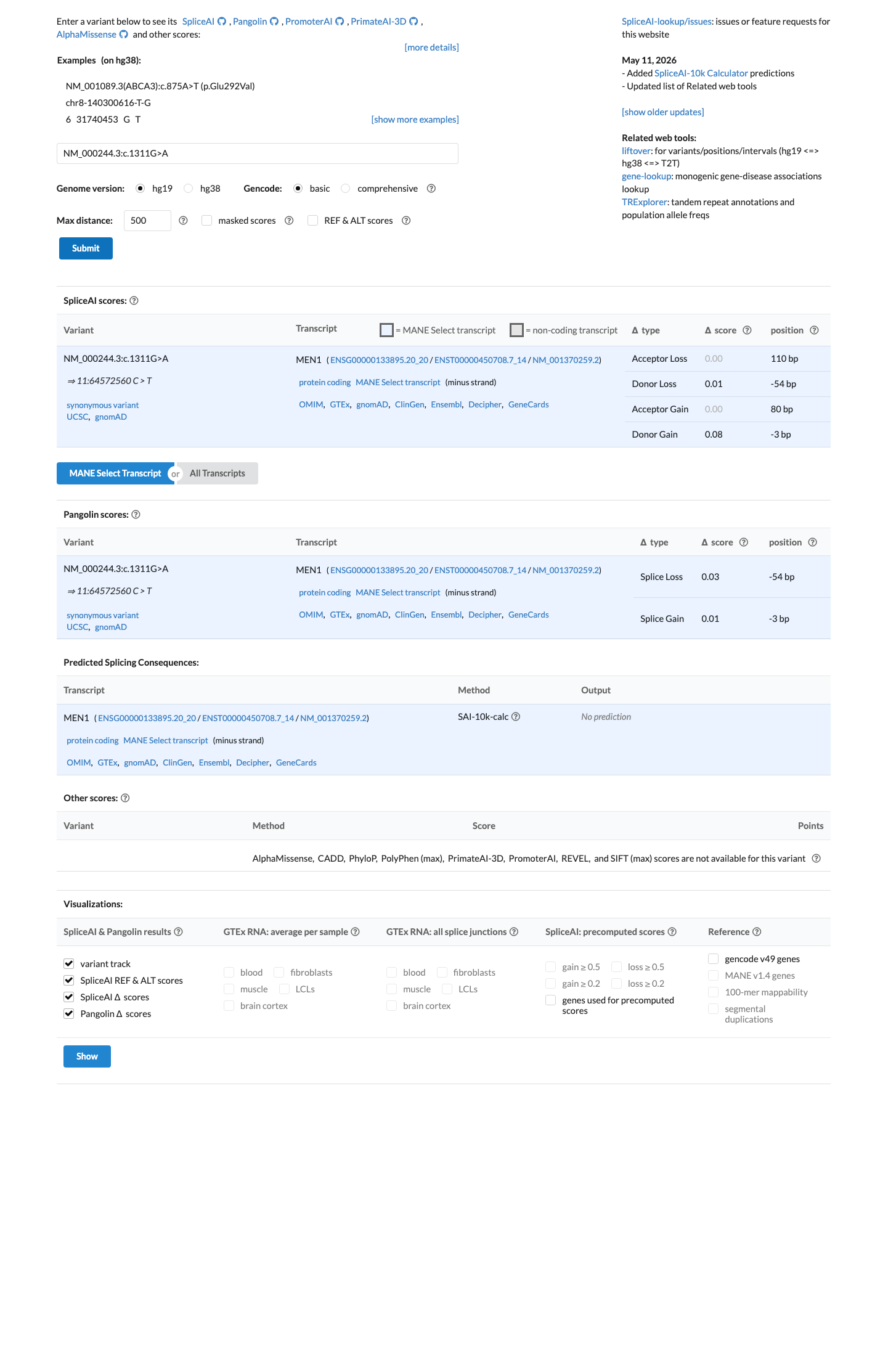
The MEN1 NM_000244.3:c.1311G>A (NP_000235.2:p.(Leu437=); NP_000235.2:p.(L437=)) variant has been reported in ClinVar predominantly as likely benign or benign, with 10 likely benign, 7 benign, and 1 uncertain significance submissions.1 This variant is present in gnomAD at 0.11182% in v2.1 and 0.14503% in v4.1, with a highest observed population frequency of 0.65789% in Amish individuals in v4.1, which is above the 0.3% BS1 threshold and argues against a rare pathogenic MEN1 variant.2 SpliceAI predicts no significant splice impact for this synonymous change, with a maximum delta score of 0.08, supporting a benign computational interpretation.3
BS1 + BP7
→
Likely Benign