Classification rationale
BS1BP4
Likely Benign
MET c.110T>C
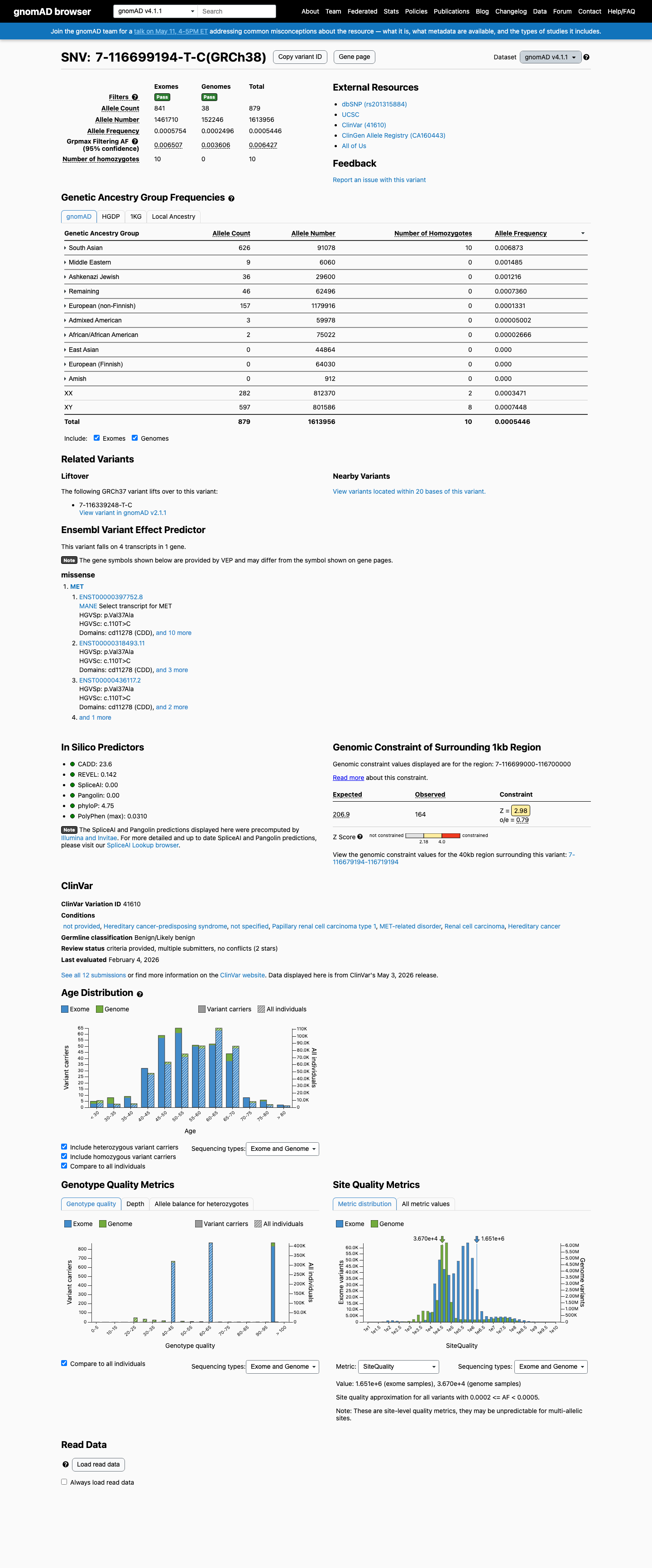
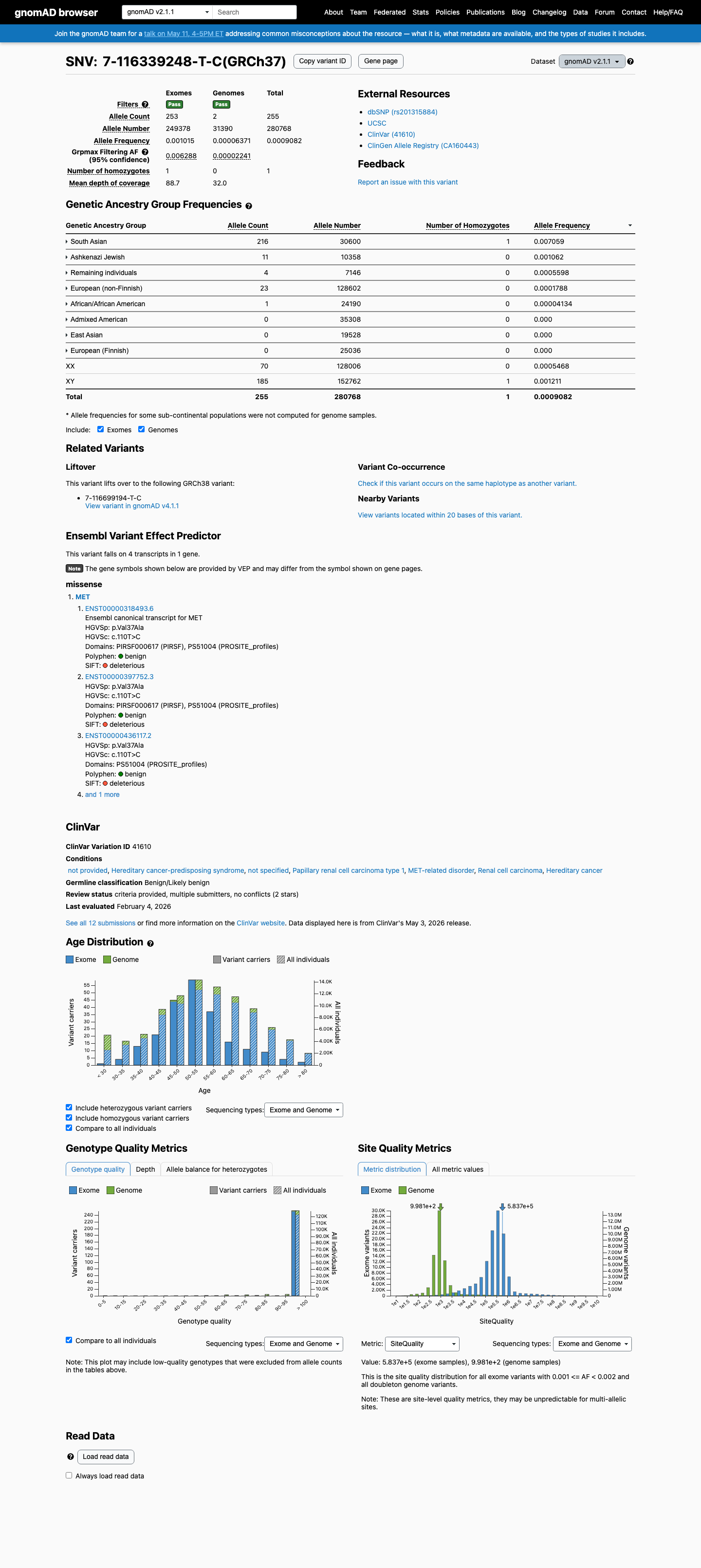
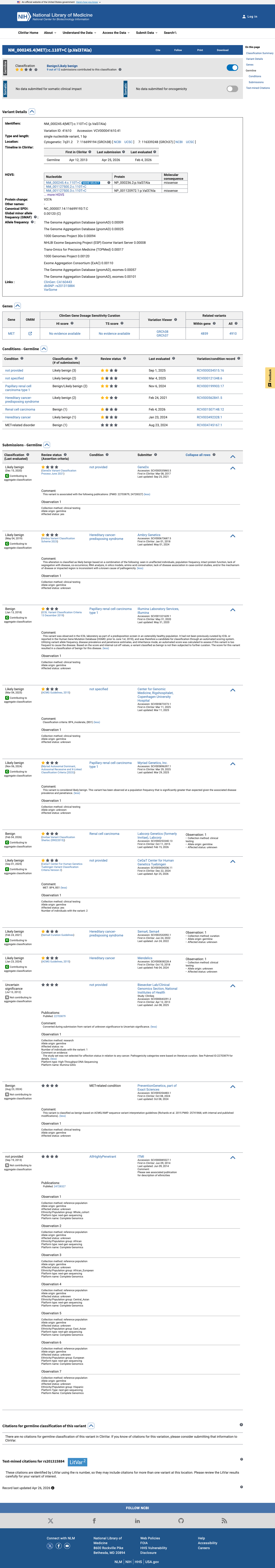
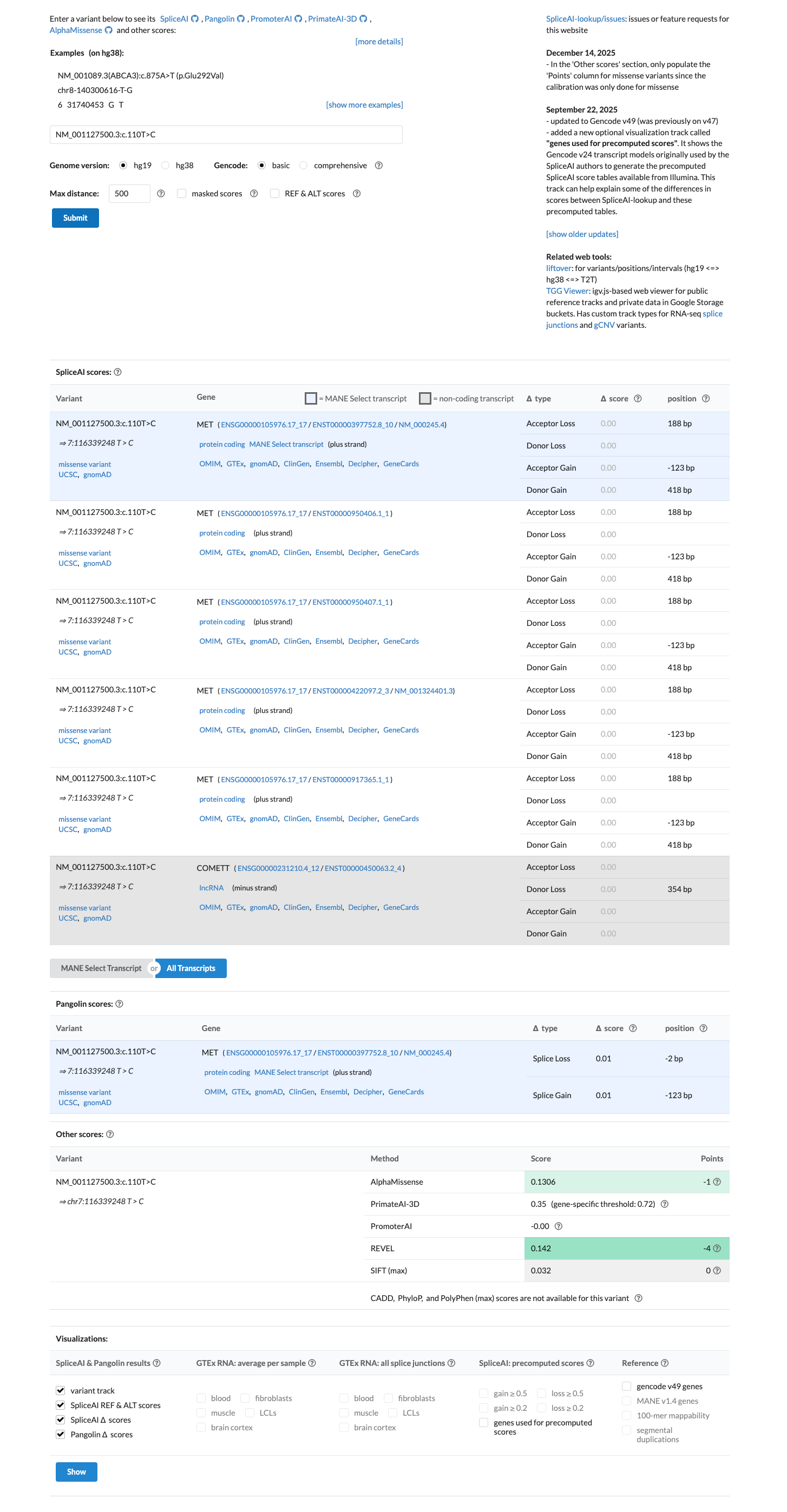
The MET c.110T>C (p.Val37Ala) variant has been reported in ClinVar with likely benign and benign submissions.1 This variant is present in population databases at a frequency above the BS1 threshold, with the highest observed frequency of 0.70588% in South Asian individuals in gnomAD v2.1 and 0.68732% in South Asian individuals in gnomAD v4.1, arguing against a rare pathogenic germline variant.2 No well-established variant-specific functional study was identified to support either a damaging or a normal effect.3 Computational evidence supports a benign interpretation, with SpliceAI predicting no significant splice impact (maximum delta score 0.00), REVEL of 0.142, and BayesDel of -0.247812.4
BS1 + BP4
→
Likely Benign