This variant is a missense substitution (NM_001128425.2:c.1255G>A, p.Ala419Thr) in exon 13 of MUTYH. The variant is present at very low frequency in gnomAD v2.1 (AF=0.00672%, 19/282,560 alleles, 0 homozygotes) and v4.1 (AF=0.00874%, 141/1,614,058 alleles, 1 homozygote), and is absent from gnomAD-Canada, meeting PM2 at the supporting level.1 Multiple in silico tools predict no damaging effect: REVEL score 0.414 (below the 0.5 pathogenic threshold), BayesDel score -0.184886 (benign-leaning), and SpliceAI delta score 0.00 (no predicted splice impact), meeting BP4 at the supporting level.2 No functional studies, segregation data, de novo observations, or variant-specific literature have been identified for this variant. The MUTYH VCEP (InSiGHT Hereditary Colorectal Cancer/Polyposis Specification v1.0.0) framework was present but incomplete with no gene-specific criteria rules; generic ACMG/AMP 2015 criteria were applied.3 In ClinVar, the variant is reported as Uncertain significance by 11 of 14 submitters. Ambry Genetics classifies as Benign and Labcorp/Invitae as Likely benign, though the underlying evidence is not publicly accessible. The single homozygote in gnomAD v4.1 warrants attention for a recessive disorder but lacks phenotype data.4 The only applicable criteria are PM2_supporting (pathogenic supporting) and BP4_supporting (benign supporting). These oppose each other, resulting in conflicting evidence insufficient to classify beyond Variant of Uncertain Significance (VUS).
MUTYH
Final classification
VUS
MUTYH c.1255G>A · p.Ala419Thr
MUTYH
This variant is a missense substitution (NM_001128425.2:c.1255G>A, p.Ala419Thr) in exon 13 of MUTYH.
InSiGHT Hereditary Colorectal Cancer/Polyposis Specification v1.0.0 lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP4
VUS
MUTYH c.1255G>A
PM2 + BP4
→
VUS
Gene diagram
· NM_001128425.2 · variants mapped to exon structure
MUTYH
NM_001128425.2
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 20 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is absent from gnomAD-Canada and present at very low frequency in gnomAD v2.1 (AF=0.00672%, 19/282,560 alleles, 0 homozygotes) and v4.1 (AF=0.00874%, 141/1,614,058 alleles, 1 homozygote). Both frequencies are well below the 0.1% PM2 threshold. The single homozygote in v4.1 is noted but does not negate PM2 at the supporting level given the overall rarity.
gnomAD v2.1: AF=0.00672%19/282560 alleles
✓
BP4
supporting
Benign
Multiple lines of computational evidence suggest this variant does not impact protein function. REVEL score is 0.414 (below the 0.5 pathogenic threshold). BayesDel score is -0.184886 (negative scores indicate benign prediction). SpliceAI predicts no splice impact (max delta = 0.00). ClinVar submitters note that multiple in silico tools predict a benign effect (e.g., 'three of five in-silico tools predict a benign effect'; 'computational prediction suggests that this variant may not impact protein structure and function'). The convergent in silico evidence supports BP4.
REVEL: 0.414 (below 0.5 threshold).BayesDel: -0.184886 (benign-leaning).SpliceAI: max delta 0.00 (no splice impact).
Assessed · not applied
Pathogenic
PS1
No evidence that the same amino acid change (p.Ala419Thr) has been established as pathogenic in a different nucleotide change in MUTYH.
PS2
No de novo observation reported for this variant.
PS3
No variant-specific functional studies have been reported.
PS4
The variant is present in gnomAD at low frequency (v2.1: AF=0.0067%, 19/282,560; v4.1: AF=0.0087%, 141/1,614,058 with 1 homozygote), which argues against significant enrichment in affected individuals.
PM1
This variant is located at residue 419 (p.Ala419Thr), which does not lie in a statistically significant somatic or germline hotspot.
PM6
No de novo observation reported for this variant.
PP1
No segregation data available.
PP2
PP2 applies to genes where missense variants are a well-established mechanism of disease and the gene has a low rate of benign missense variation.
PP3
In silico predictions are mixed and do not support a deleterious effect.
PP4
PP4 requires a highly specific phenotype or family history consistent with a single genetic etiology.
PP5
PP5 requires that a reputable source (e.g., expert clinical laboratory with strong reputation) has recently classified the variant as pathogenic/likely pathogenic with supporting evidence not available to this assessment.
Benign
BA1
The variant frequency in gnomAD is well below the 1% BA1 threshold (v2.1: AF=0.0067%; v4.1: AF=0.0087%).
BS1
The variant frequency in gnomAD is below the 0.3% BS1 threshold (v2.1: AF=0.0067%; v4.1: AF=0.0087%).
BS2
BS2 requires observation in a healthy adult homozygous for a recessive disorder with full penetrance expected at an early age.
BS3
BS3 requires well-established in vitro or in vivo functional studies showing no damaging effect on protein function or splicing.
BS4
No segregation data demonstrating lack of cosegregation with disease.
BP1
BP1 applies to missense variants in genes where only truncating variants cause disease.
BP2
BP2 requires observation in trans with a known pathogenic variant or in cis with a pathogenic variant in a dominant disorder in a healthy individual.
BP5
BP5 requires that the variant be found in a case with an alternate molecular basis for disease (e.g., a second pathogenic variant in another gene explaining the phenotype).
BP6
BP6 requires a reputable source to classify the variant as benign/likely benign with evidence not available to this assessment.
N/A · 6
PVS1 · PM3 · PM4 · PM5 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
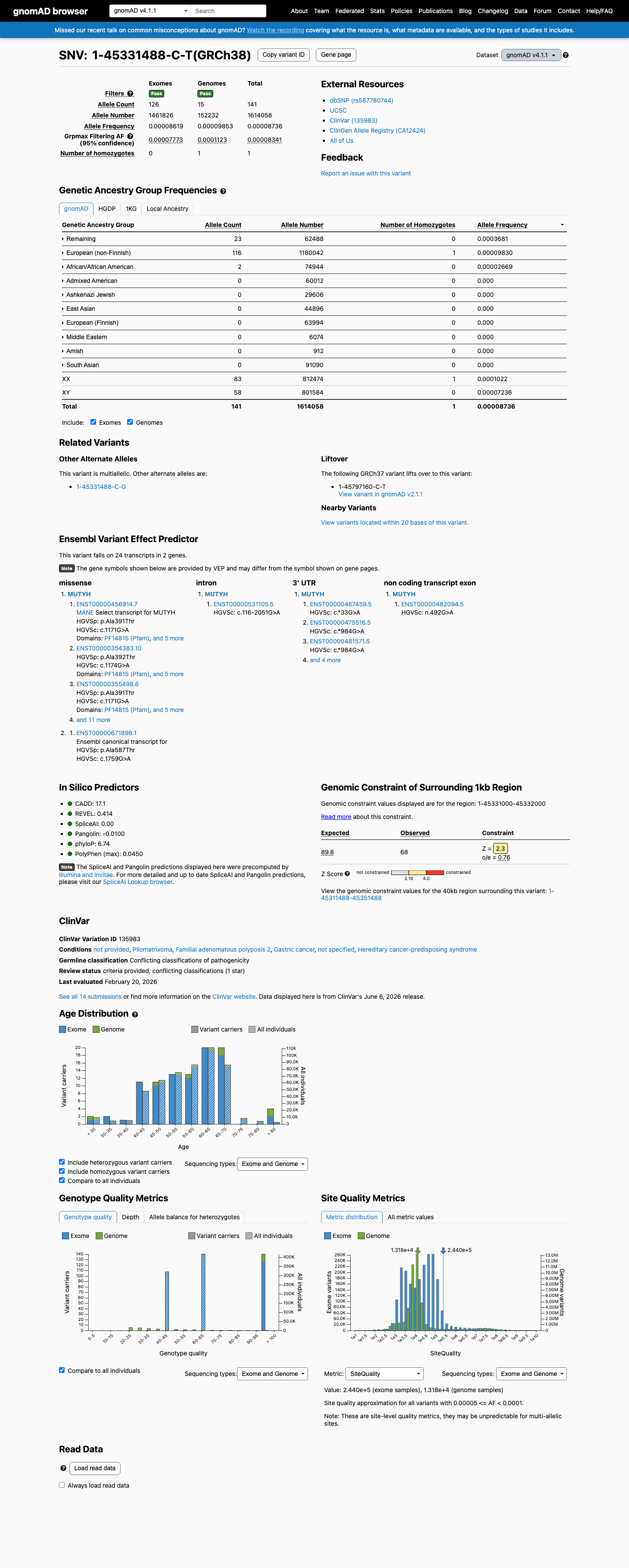
This variant is present in gnomAD v4.1 (AF= 8.73575e-05; MAF= 0.00874%, 141/1614058 alleles, homozygotes = 1) and has highest observed frequency in the Remaining individuals population (AF= 0.000368071; MAF= 0.03681%, 23/62488 alleles, homozygotes = 0); grpmax FAF= 8.341e-05.
v2.1
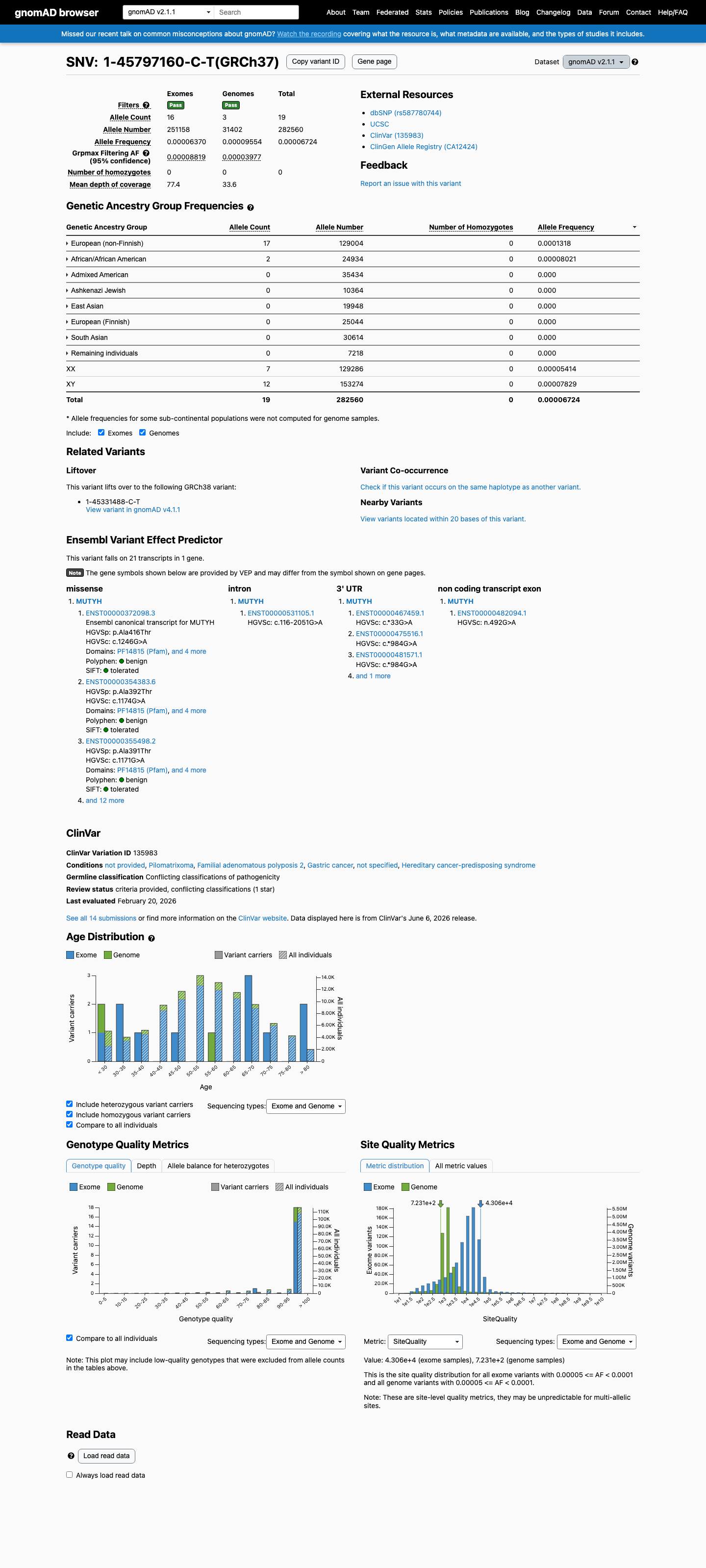
This variant is present in gnomAD v2.1 (AF= 6.72424e-05; MAF= 0.00672%, 19/282560 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 0.000131779; MAF= 0.01318%, 17/129004 alleles, homozygotes = 0); grpmax FAF= 8.819e-05.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.00016291951775822744, 3/18414 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0087%
· 141 / 1,614,058
1 hom · FAF 0.0083%
1 hom · FAF 0.0083%
Remaining individuals 23 / 62,488 |
0.037% |
European (non-Finnish) 116 / 1,180,042 |
0.0098% 1 hom |
African/African American 2 / 74,944 |
0.0027% |
+ 7 not observed (Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish)
gnomAD v2.1
0.0067%
· 19 / 282,560
0 hom · FAF 0.0088%
0 hom · FAF 0.0088%
European (non-Finnish) 17 / 129,004 |
0.013% |
African/African American 2 / 24,934 |
0.008% |
+ 6 not observed (Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
0.016%
· 3 / 18,414
0 hom · FAF 0.0069%
0 hom · FAF 0.0069%
indel · split
European (non-Finnish) 3 / 11,736 |
0.026% |
+ 8 not observed (African/African American, Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, Remaining individuals, South Asian)
ClinVar
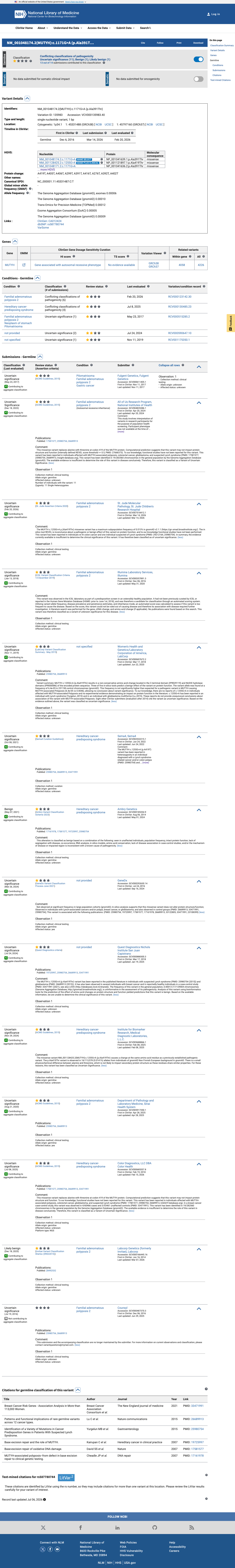
This variant has been reported in ClinVar as Uncertain significance (10 clinical laboratories) and as Uncertain Significance (1 clinical laboratory) and as Benign (1 clinical laboratory) and as Likely benign (1 clinical laboratory). (ClinVarID = 135983)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.414. BayesDel score = -0.184886.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. MUTYH, a DNA glycosylase, is frequently mutated in colorectal cancer.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
9Sources
Triaged references · 9 PMIDs not cited in assessment
23852704 ↗
Tumor markers in colorectal cancer, gastric cancer and gastrointestinal stromal cancers: European group on tumor markers 2014 guidelines update.
CLINVAR
25645574 ↗
ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
26689913 ↗
Patterns and functional implications of rare germline variants across 12 cancer types.
CLINVAR
17161978 ↗
MUTYH-associated polyposis--from defect in base excision repair to clinical genetic testing.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR