NM_006231.4:c.6531+4C>T is an intronic variant at position +4 of intron 46 in POLE. SpliceAI predicts no significant splicing impact (max delta score 0.13).1 This variant is present in gnomAD at extremely low frequency (v2.1: 8/245,768 alleles, AF=3.26e-05; v4.1: 28/1,589,258 alleles, AF=1.76e-05). The frequency does not reach BA1 (>1%) or BS1 (>0.3%) thresholds.2 Invitae (Labcorp Genetics) reports this variant as Likely benign with criteria provided in ClinVar (SCV000772749). Ambry Genetics reports it as Uncertain significance (SCV000671346). Both are single-submitter clinical testing classifications without expert panel review.3 The only criterion met is BP6 (Supporting benign) based on the Invitae ClinVar submission reporting Likely benign. No pathogenic criteria are met. Under ACMG/AMP 2015 combination rules, a single Supporting benign criterion is insufficient for a Likely benign classification (requires ≥2 Supporting benign or 1 Strong benign + 1 Supporting benign). The variant is classified as a Variant of Uncertain Significance (VUS).4
POLE
Final classification
VUS
POLE c.6531+4C>T · p.?
POLE
NM_006231.4:c.6531+4C>T is an intronic variant at position +4 of intron 46 in POLE. SpliceAI predicts no significant splicing impact (max delta score 0.13).
Only one criterion met: BP6 (supporting benign) based on Invitae ClinVar submission reporting Likely benign. No pathogenic criteria are met. Under the León-Castillo et al. 2020 custom POLE framework (which mirrors ACMG/AMP 2015 final combination rules), a single supporting benign criterion is insufficient for Likely Benign (requires ≥2 Supporting benign or 1 Strong benign + 1 Supporting benign). The variant defaults to Variant of Uncertain Significance (VUS).
Classification rationale
BP6
VUS
POLE c.6531+4C>T
BP6
→
VUS
Gene diagram
· NM_006231.4 · variants mapped to exon structure
POLE
NM_006231.4
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 17 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
BP6
supporting
Benign
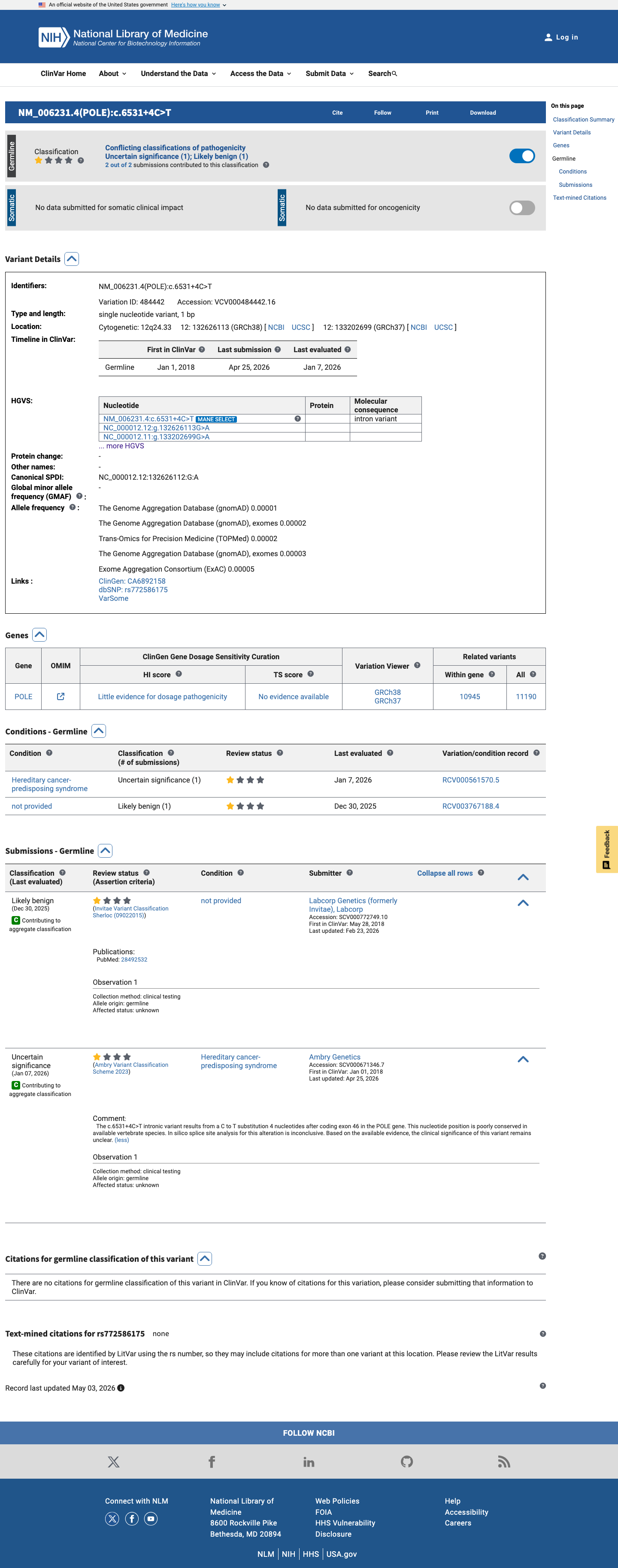
Invitae (Labcorp Genetics), a reputable clinical testing laboratory, reports this variant as Likely benign with criteria provided in ClinVar (SCV000772749, review status: criteria provided, single submitter). Under generic ACMG/AMP, a reputable source reporting the variant as benign supports BP6 at the Supporting level.
ClinVar SCV000772749: Likely benign from Invitae (Labcorp Genetics)criteria providedReputable clinical laboratory with criteria-based assessment
Assessed · not applied
Pathogenic
PVS1
Intronic variant at c.6531+4 (intron 46), outside the canonical ±1,2 splice consensus.
PS2
No de novo data available for this variant.
PS4
Two single-submitter ClinVar entries (Likely benign from Invitae, Uncertain significance from Ambry) do not constitute significantly increased prevalence in affected individuals.
PM2
Variant is present in gnomAD at extremely low frequency (v2.1: 8/245,768 alleles, AF=3.26e-05; v4.1: 28/1,589,258 alleles, AF=1.76e-05) and SpliceAI predicts no splicing impact.
PM6
No de novo data available for this variant.
PP1
No segregation data available for this variant.
PP3
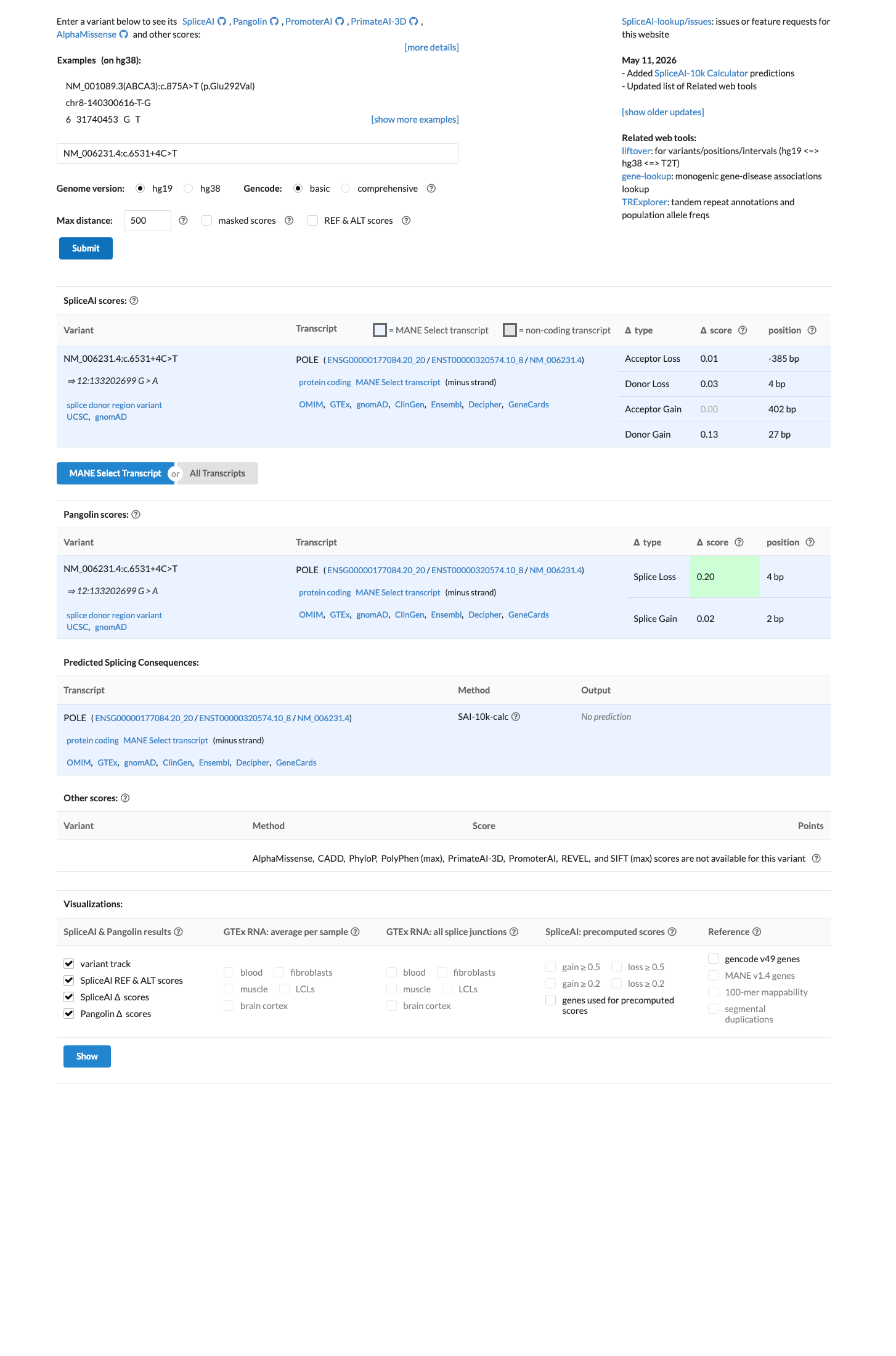
SpliceAI predicts no significant splicing impact (max delta 0.13).
PP4
No patient phenotype or family history data are available to assess whether the phenotype is highly specific for a POLE-related disorder.
PP5
ClinVar reports this variant as Likely benign (Invitae, 1 submitter) and Uncertain significance (Ambry, 1 submitter).
Benign
BA1
gnomAD v2.1 allele frequency = 3.26e-05 (0.003%) and gnomAD v4.1 = 1.76e-05 (0.002%), both well below the 1% BA1 threshold for a dominant disorder.
BS1
The variant frequency (gnomAD v2.1 AF=3.26e-05, 0.003%; v4.1 AF=1.76e-05, 0.002%) is well below the 0.3% BS1 threshold.
BS2
No data on observation of this variant in healthy adult individuals for a disorder with full penetrance expected at an early age.
BS3
No well-established in vitro or in vivo functional studies are available for this variant.
BS4
No segregation data available to evaluate lack of segregation in affected family members.
BP2
No data on observation of this variant in trans with a pathogenic variant for a dominant disorder or in cis with a pathogenic variant.
BP4
SpliceAI predicts no significant splicing impact (max delta 0.13), but this constitutes only a single line of computational evidence.
BP5
No data on observation of this variant in a case with an alternate molecular basis for disease.
N/A · 10
PS1 · PS3 · PM1 · PM3 · PM4 · PM5 · PP2 · BP1 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
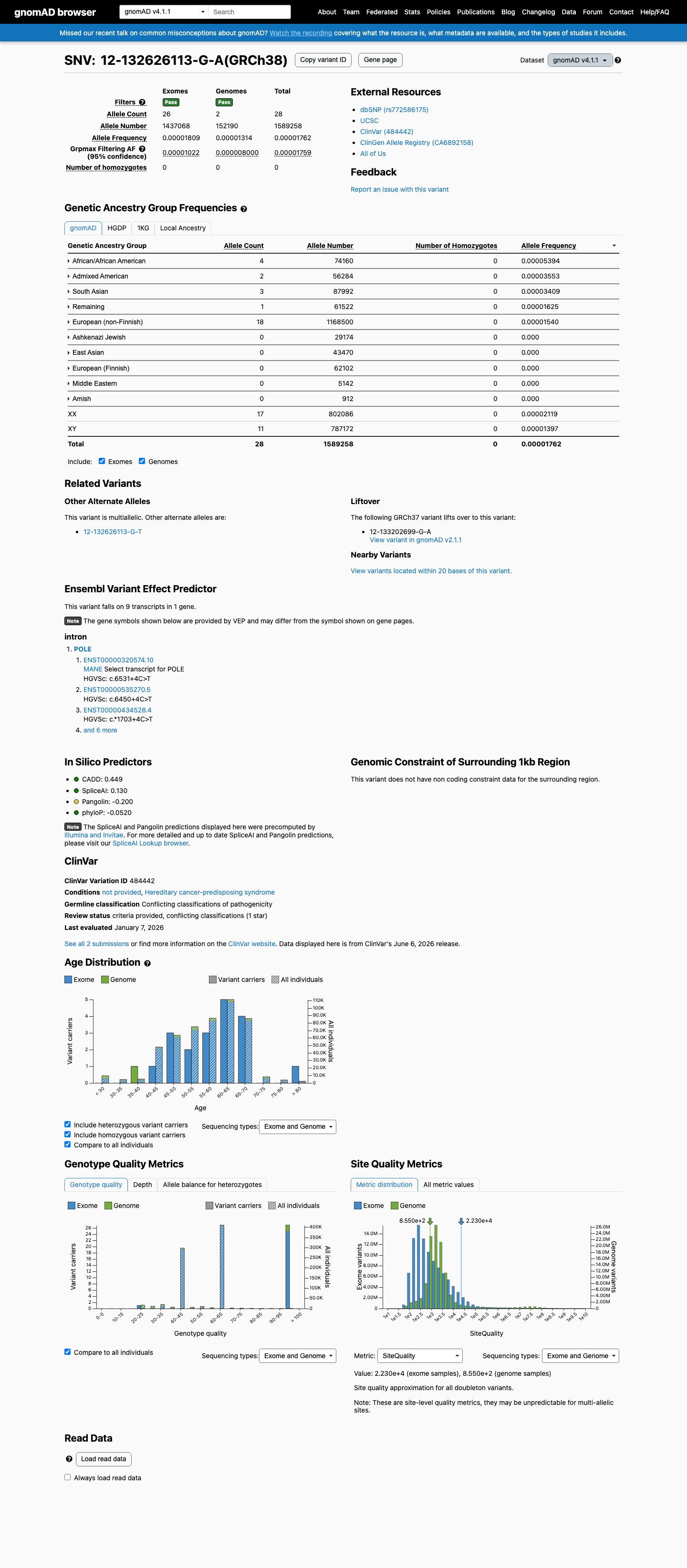
This variant is present in gnomAD v4.1 (AF= 1.76183e-05; MAF= 0.00176%, 28/1589258 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 5.39374e-05; MAF= 0.00539%, 4/74160 alleles, homozygotes = 0); grpmax FAF= 1.759e-05.
v2.1
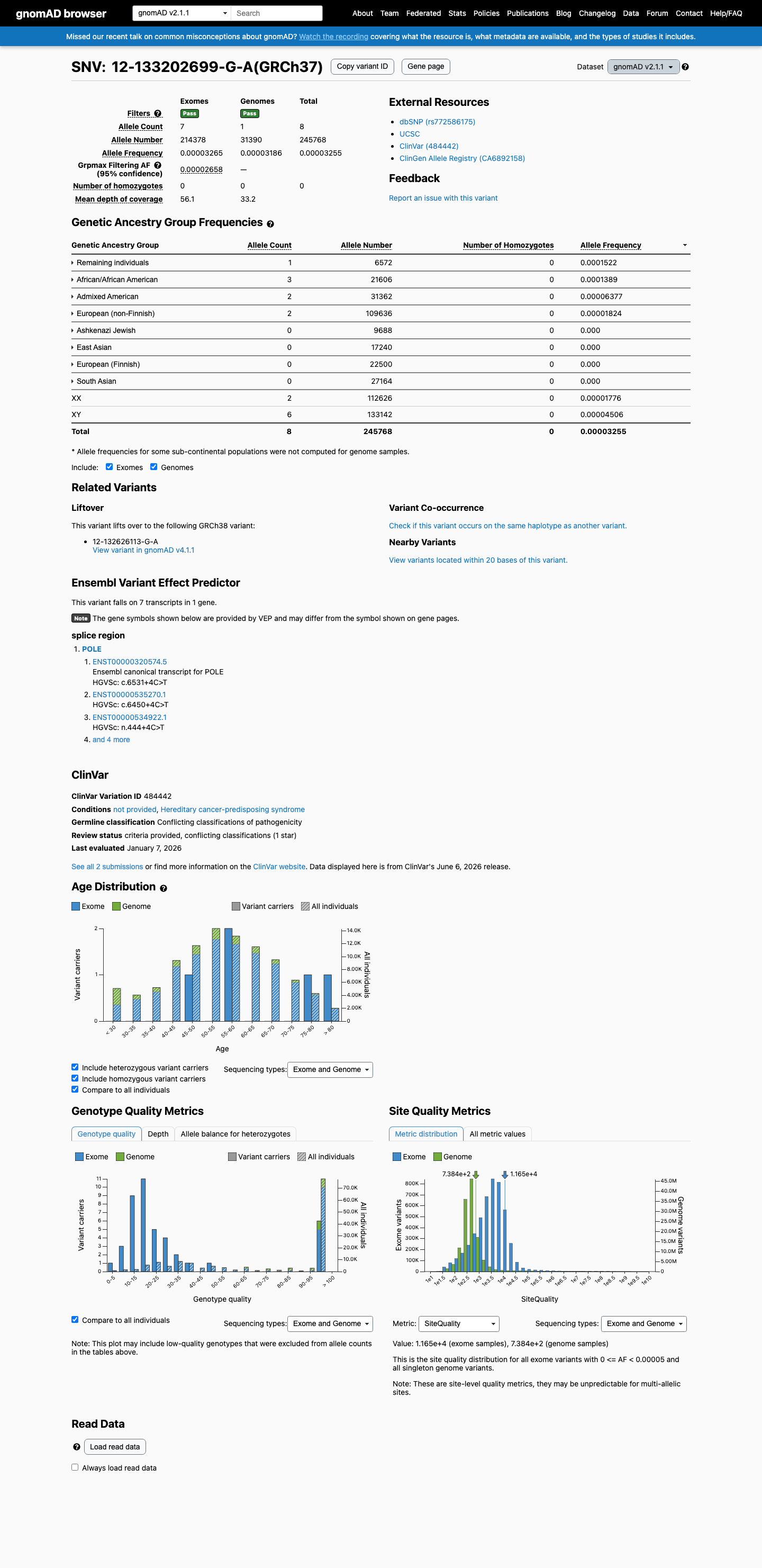
This variant is present in gnomAD v2.1 (AF= 3.2551e-05; MAF= 0.00326%, 8/245768 alleles, homozygotes = 0) and has highest observed frequency in the Remaining individuals population (AF= 0.000152161; MAF= 0.01522%, 1/6572 alleles, homozygotes = 0); grpmax FAF= 2.658e-05.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0018%
· 28 / 1,589,258
0 hom · FAF 0.0018%
0 hom · FAF 0.0018%
African/African American 4 / 74,160 |
0.0054% |
Admixed American 2 / 56,284 |
0.0036% |
South Asian 3 / 87,992 |
0.0034% |
Remaining individuals 1 / 61,522 |
0.0016% |
European (non-Finnish) 18 / 1,168,500 |
0.0015% |
+ 5 not observed (European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.0033%
· 8 / 245,768
0 hom · FAF 0.0027%
0 hom · FAF 0.0027%
Remaining individuals 1 / 6,572 |
0.015% |
African/African American 3 / 21,606 |
0.014% |
Admixed American 2 / 31,362 |
0.0064% |
European (non-Finnish) 2 / 109,636 |
0.0018% |
+ 4 not observed (Ashkenazi Jewish, East Asian, European (Finnish), South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Likely benign (1 clinical laboratory) and as Uncertain significance (1 clinical laboratory). (ClinVarID = 484442)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.13).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
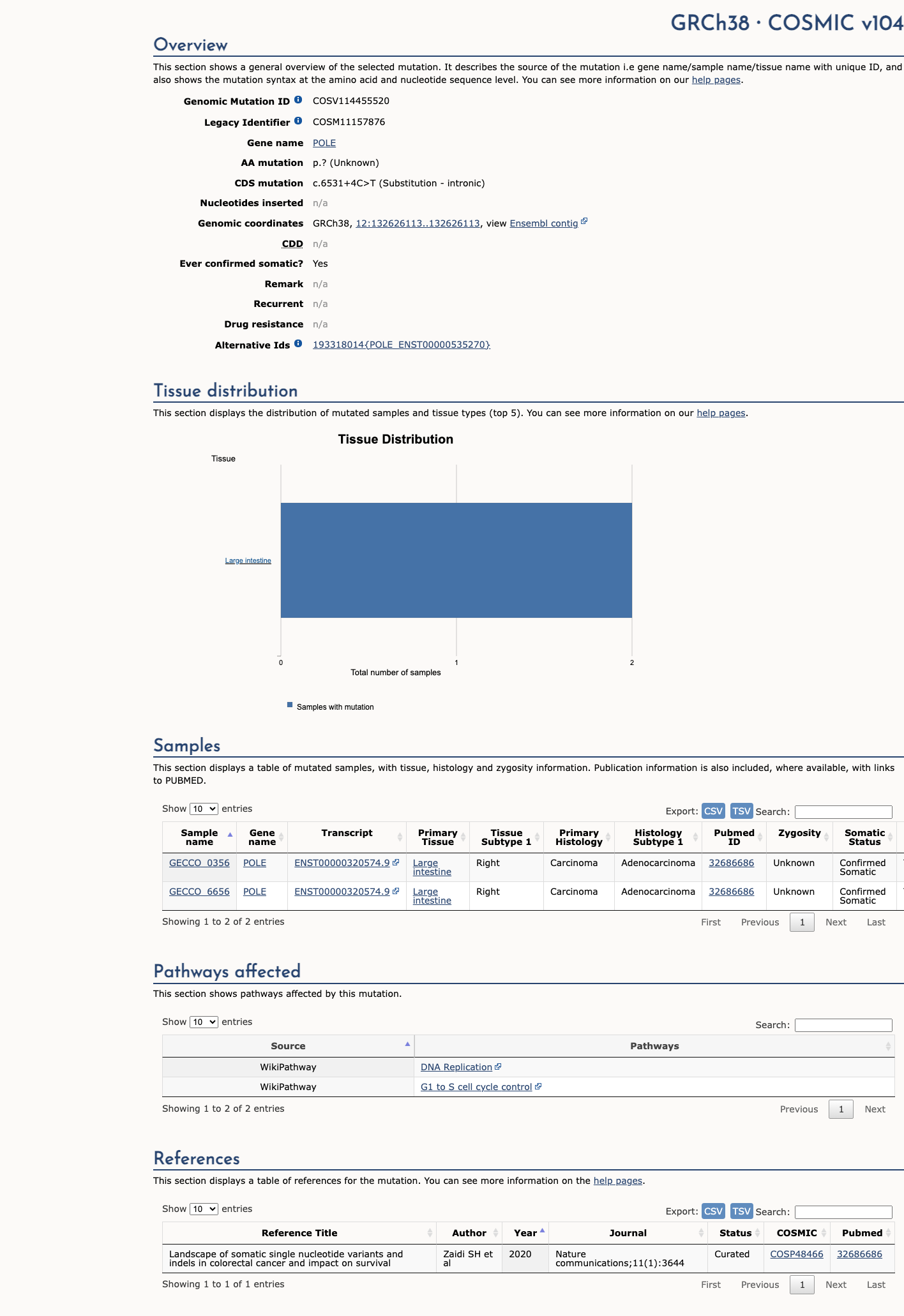
COSMIC
Somatic evidence
COSMIC
This variant has previously been reported in somatic cancers (COSMIC; COSV114455520, n = 2 times).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 2 PMIDs not cited in assessment
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR